Constructing microbial cell factories for monoterpene natural products
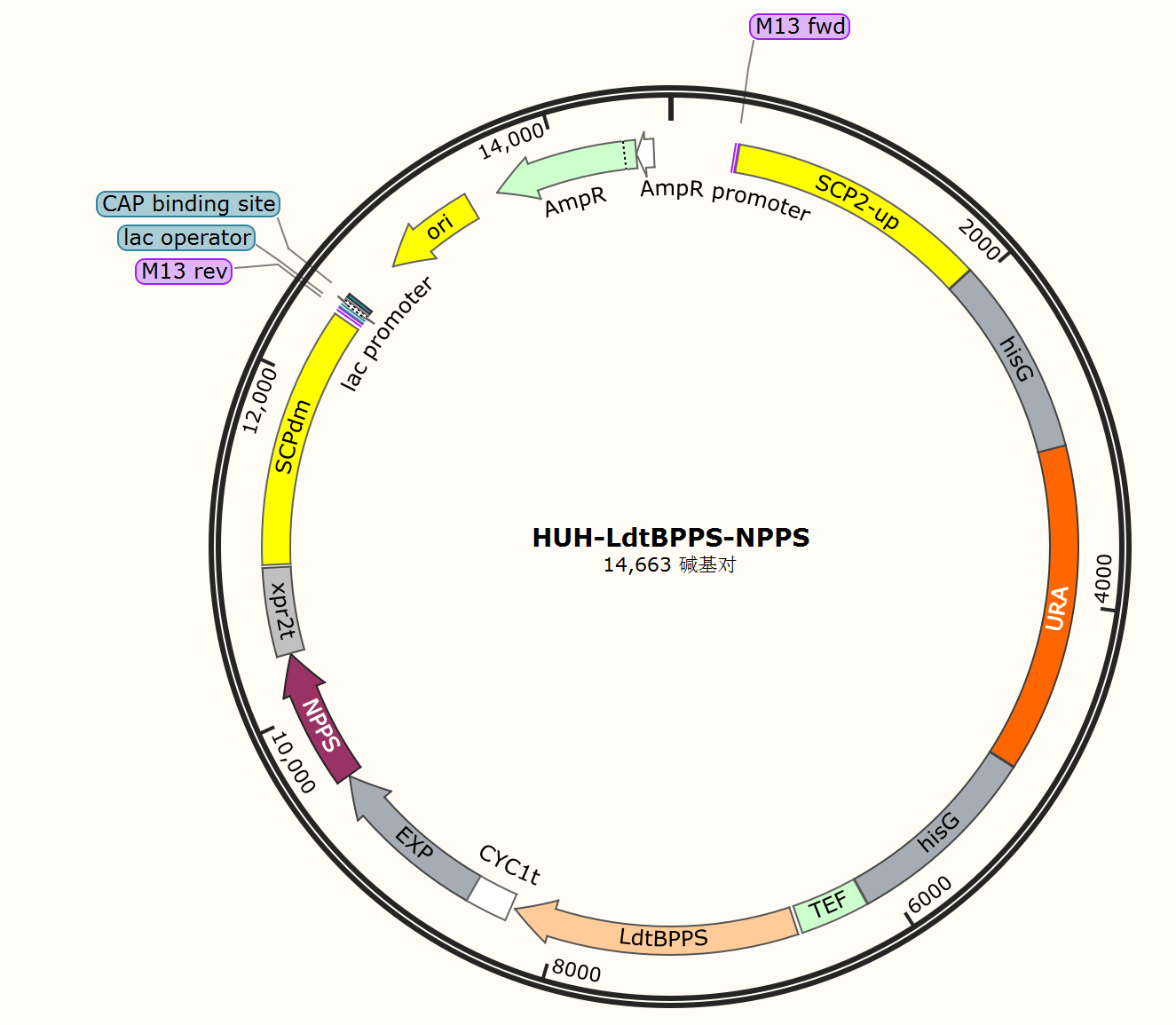
To synthesize nerol, NPP serves as the precursor and the pathway involves a two-step reaction of DMAPP and IPP to generate NPP. Subsequently, dephosphorylation of NPP leads to the formation of nerol. The first step is catalyzed by NPP synthase (SlNPPS) obtained from Solanum lycopersicum, while the second step is facilitated by nerol synthase LdtBPPSF162A. To this end, we constructed the pHUH-LdtBPPS-NPPS plasmid and introduced it into the PO1f strain, resulting in the generation of a nerol production strain, PLN1.
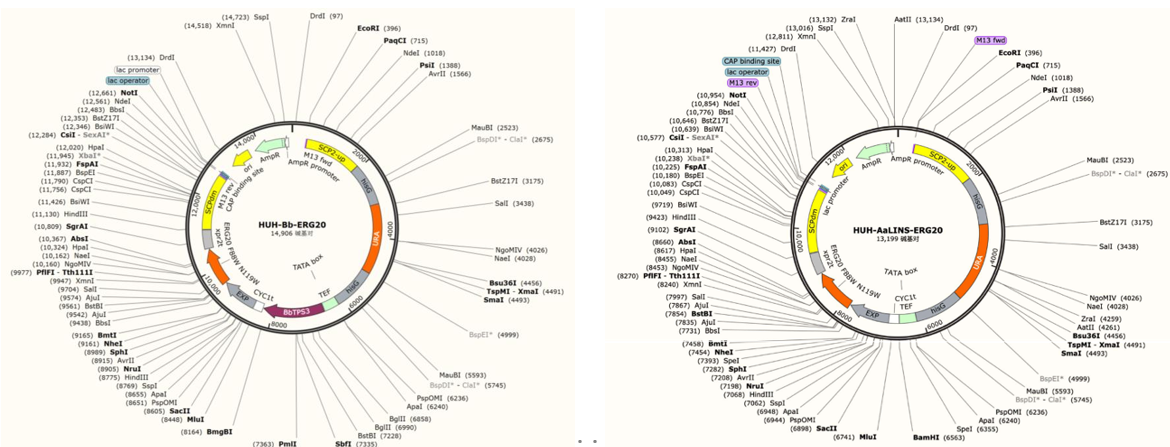
For the synthesis of borneol and linalool, GPP acts as the precursor. Within the metabolic process of the MVA pathway, the bifunctional enzyme ERG20 plays a crucial role in catalyzing the synthesis of both GPP and farnesyl pyrophosphate (FPP). To enhance the intracellular concentration of GPP, we employed site-directed mutagenesis to introduce double mutations (F88W and N119W) into the ERG20 enzyme. This modification reduced the activity of FPP synthase, thereby increasing the availability of the monoterpene precursor, GPP. Consequently, we constructed the pHUH-BbTPS3-ERG20 and pHUH-AaLINS-ERG20 plasmids utilizing the BbTPS3 gene and AaLINS gene along with the ERG20F88WN119W mutations. These plasmids were then introduced into the PO1f strain, resulting in the successful creation of the borneol production strain, PBE1, and the linalool production strain, PAE1.
Following the fermentation process, we measured the concentration of borneol in the culture of PBE1 through GC-MS analysis. The molecular weight of borneol is 154. However, after searching for MS fragments at all elution times, we were unable to detect any molecular ion peak with a relative molecular weight of 154 in the fermentation product of PBE1. This suggests that borneol was not detected in the sample analyzed, and the elution peak observed in the GC results was not the intended target product.
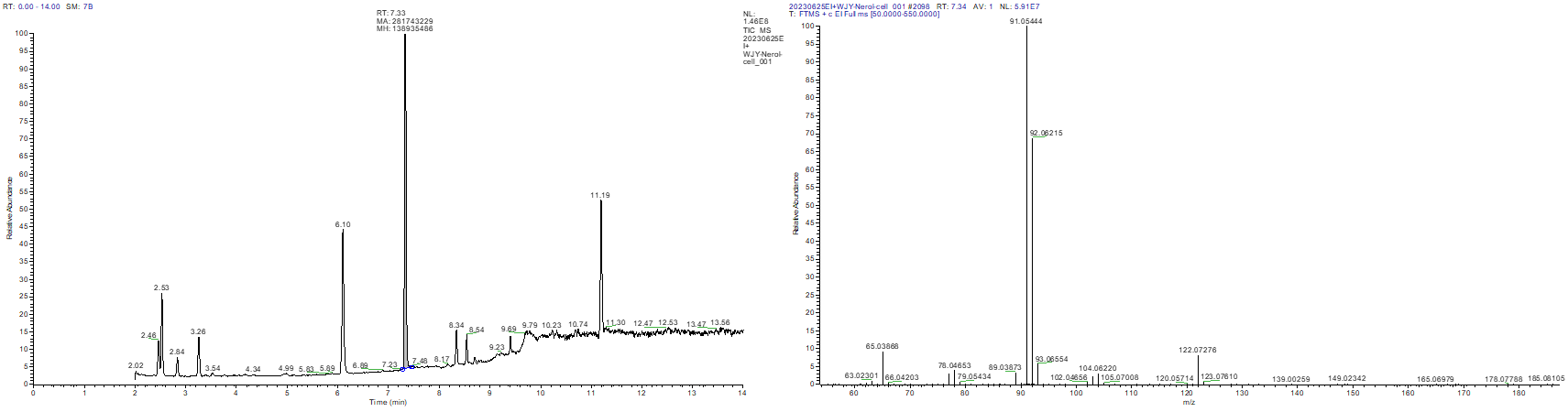
Fig.1. Plasmid maps of pHUH-LdtBPPS-NPPS constructed in this part.
Fig.2. Plasmid maps of pHUH-BbTPS3-ERG20 and pHUH-AaLINS-ERG20 constructed in this part.
Following the fermentation process, we measured the concentration of borneol in the culture of PBE1 through GC-MS analysis. The molecular weight of borneol is 154. However, after searching for MS fragments at all elution times, we were unable to detect any molecular ion peak with a relative molecular weight of 154 in the fermentation product of PBE1. This suggests that borneol was not detected in the sample analyzed, and the elution peak observed in the GC results was not the intended target product.
Fig.3. GC-MS detection of the production status of borneol. (A) Elution results of PBE1 fermentation product in the first 14 minutes of GC analysis; (B) Mass spectrometry results of possible elution time for borneol.
Successfully establishing circular monoterpene biosensors in Y. lipolytica
After preliminary experiments, it was found that the constructed promoters with the 5th to the 8th insertion sites had no induction activity, that is, there was no difference in fluorescence intensity before and after borneol induction. Therefore, the yeast containing the inducible promoter of the first to fourth insertion sites was cultured, and the expression of hrGFP was induced using a medium containing 0 mM, 0.5 mM, 1 mM, and 2 mM borneol. The yeast containing the inducible promoter of the fifth insertion site was cultured as a negative control. After 22 hours of culture, the relative fluorescence intensity (RFU) and OD600 were detected by microplate reader, and the ratio of RFU/OD600 was the expression intensity of yeast green fluorescent protein. In the induction experiment, the second, third and fourth insertion positions made the promoter show a significant borneol induction effect, and the second and third insertion positions showed a good positive correlation with the concentration of borneol in a certain concentration range.
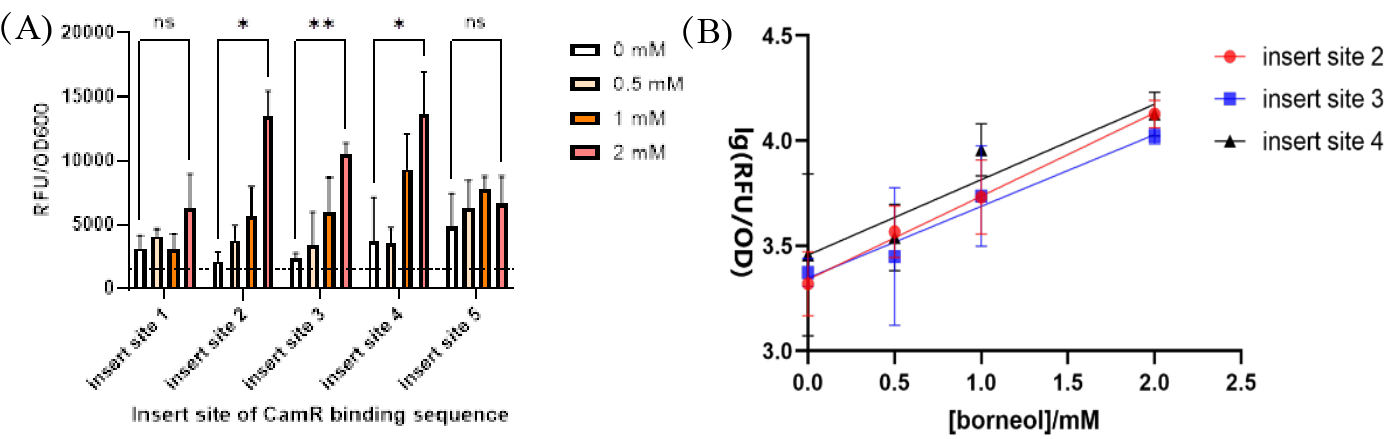
Notably, without the addition of borneol, each group showed a certain fluorescence intensity, indicating that the binding of CamR to the pTEF1 promoter could not completely inhibit the expression of the reporter gene, and there was still a certain leakage. In addition, the insertion of the CamR binding sequence may destroy part of the promoter structure, resulting in a decrease in the strength of the pTEF1 promoter.
According to the results of the above experiments, it can be concluded that, we can effectively construct the borneol-induced promoter in Y. lipolytica, in the case of that the CamR binding sequence is inserted into the pTEF1 promoter and the CamR operon protein is expressed; the induction efficiency of the promoter is related to the insertion site of the CamR binding sequence. The CamR binding sequence was inserted into the pTEF1 promoter at the second and third insertion sites, and co-expressed with the CamR gene, which has the potential to be developed as a Y. lipolytica borneol biosensor. Furthermore, That provides a highly effective tool for high-throughput screening of directed evolution of terpene synthases.
Fig.4. The induction effect of different concentrations of borneol on the constructed borneol-induced promoter. (A) The average value of RFU / OD600 in the blank control group was 1508.37, which was marked with a black dotted line in the figure. In the case of 2 mM borneol induction, the insertion positions 2,3,4 were significantly different from the non-induced group (P < 0.05), and the insertion position 3 reached a very significant level (P < 0.01). (B) The logarithm of RFU / OD600 at insertion positions 2,3,4 [ lg (RFU / OD600)] was linearly fitted with the concentration of borneol. In the range of 0 mM ~ 2 mM, the lg (RFU / OD600) of insertion sites 2 and 3 showed a good linear relationship with the concentration of borneol (R2 > 0.9, R2 > 0.99 of insertion site 2).
According to the results of the above experiments, it can be concluded that, we can effectively construct the borneol-induced promoter in Y. lipolytica, in the case of that the CamR binding sequence is inserted into the pTEF1 promoter and the CamR operon protein is expressed; the induction efficiency of the promoter is related to the insertion site of the CamR binding sequence. The CamR binding sequence was inserted into the pTEF1 promoter at the second and third insertion sites, and co-expressed with the CamR gene, which has the potential to be developed as a Y. lipolytica borneol biosensor. Furthermore, That provides a highly effective tool for high-throughput screening of directed evolution of terpene synthases.
Constructing and testing the nyEvolvR system in K. marxianus
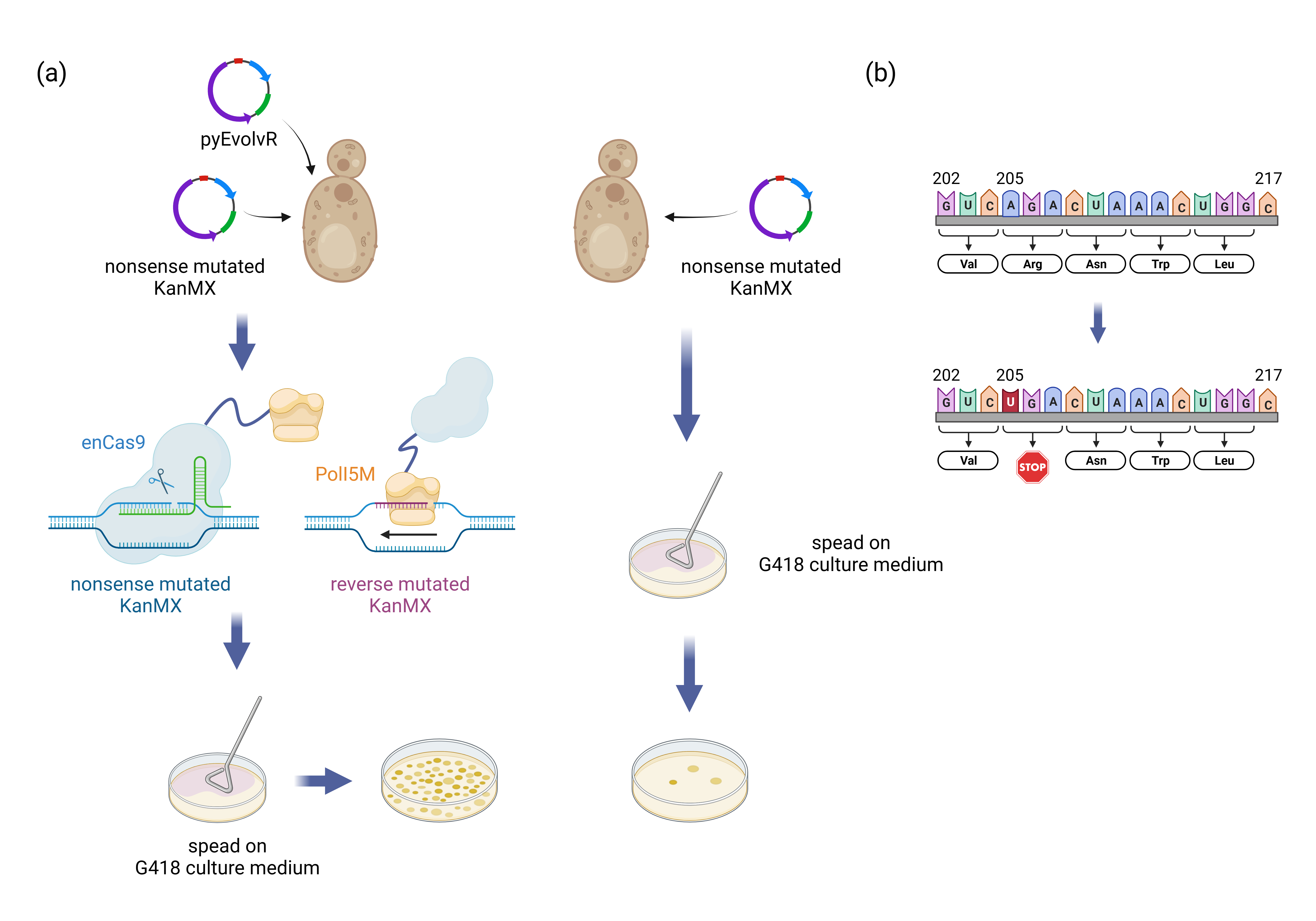
Fig.5. Process of constructing and testing the continuous directed evolution system nyEvolvR.
Next :Future Page