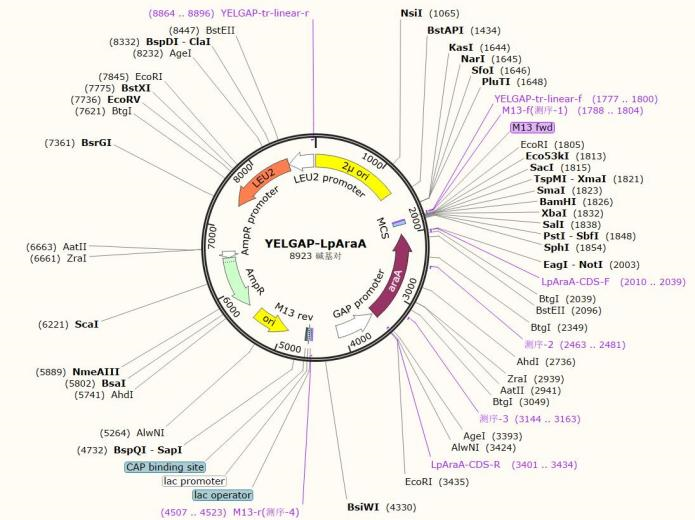
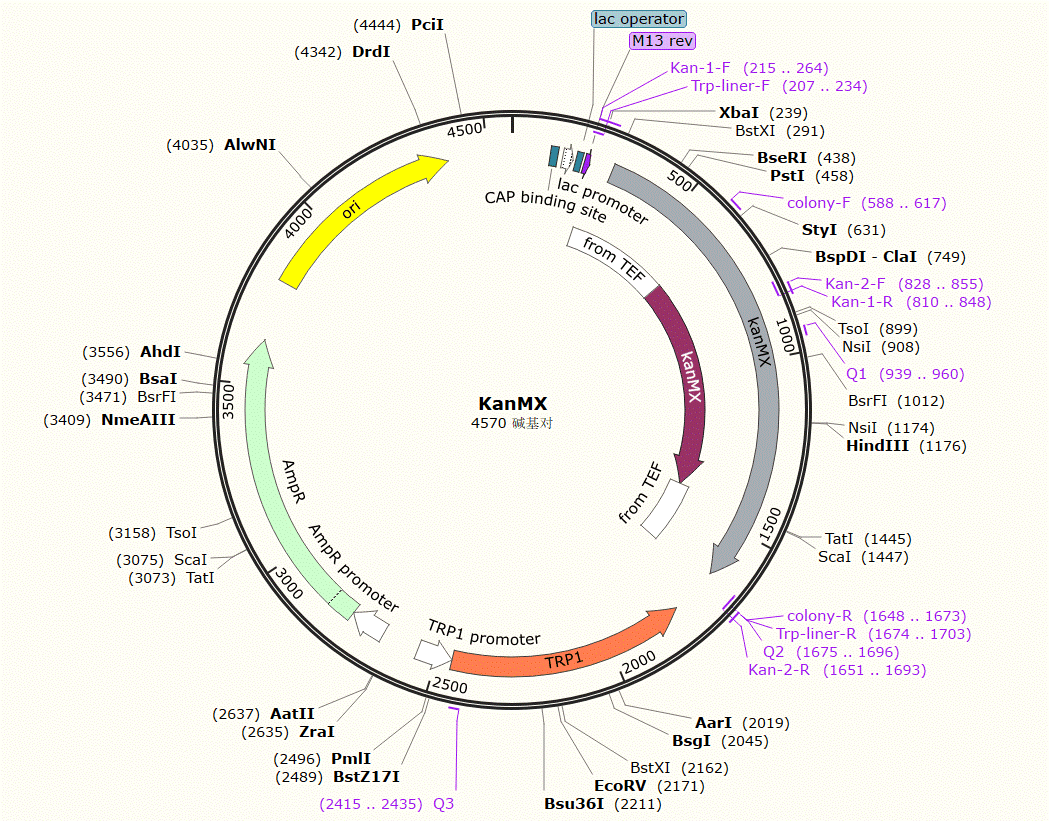
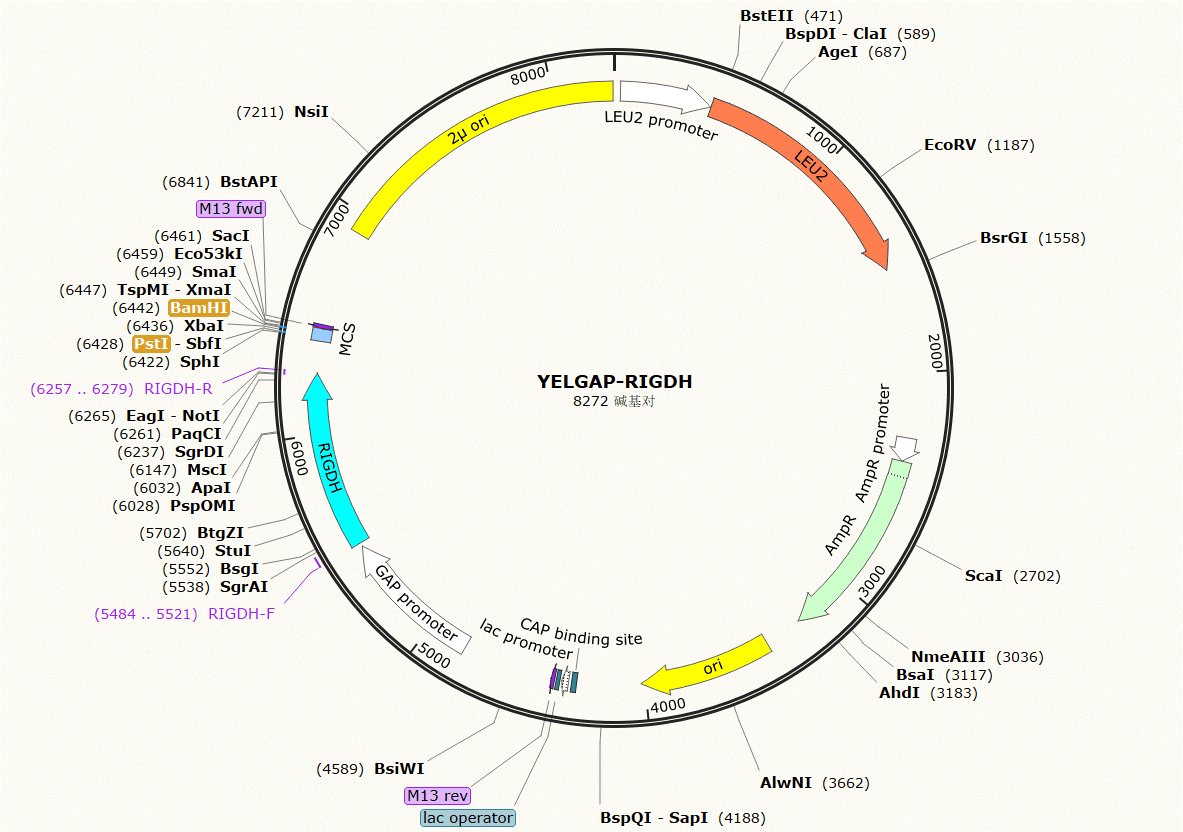
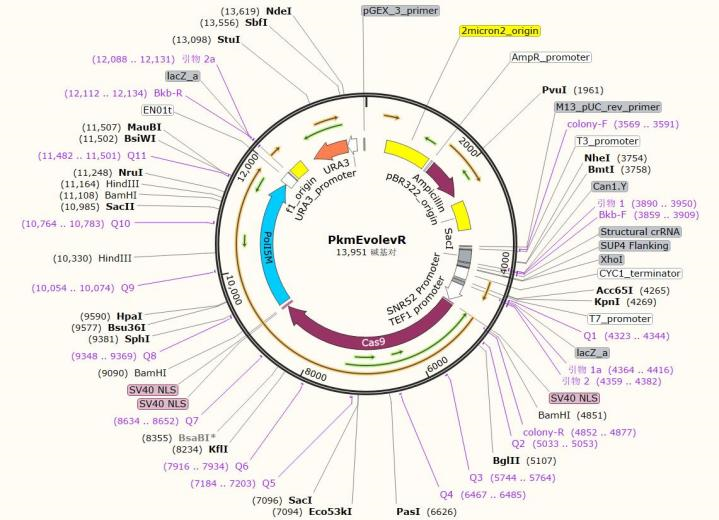
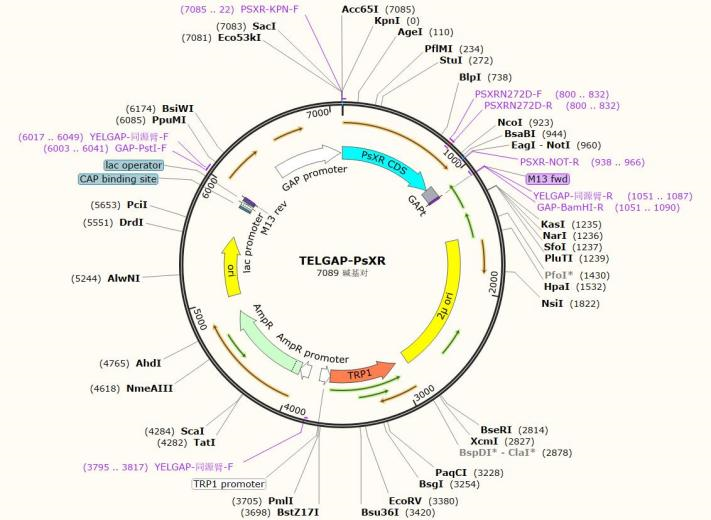
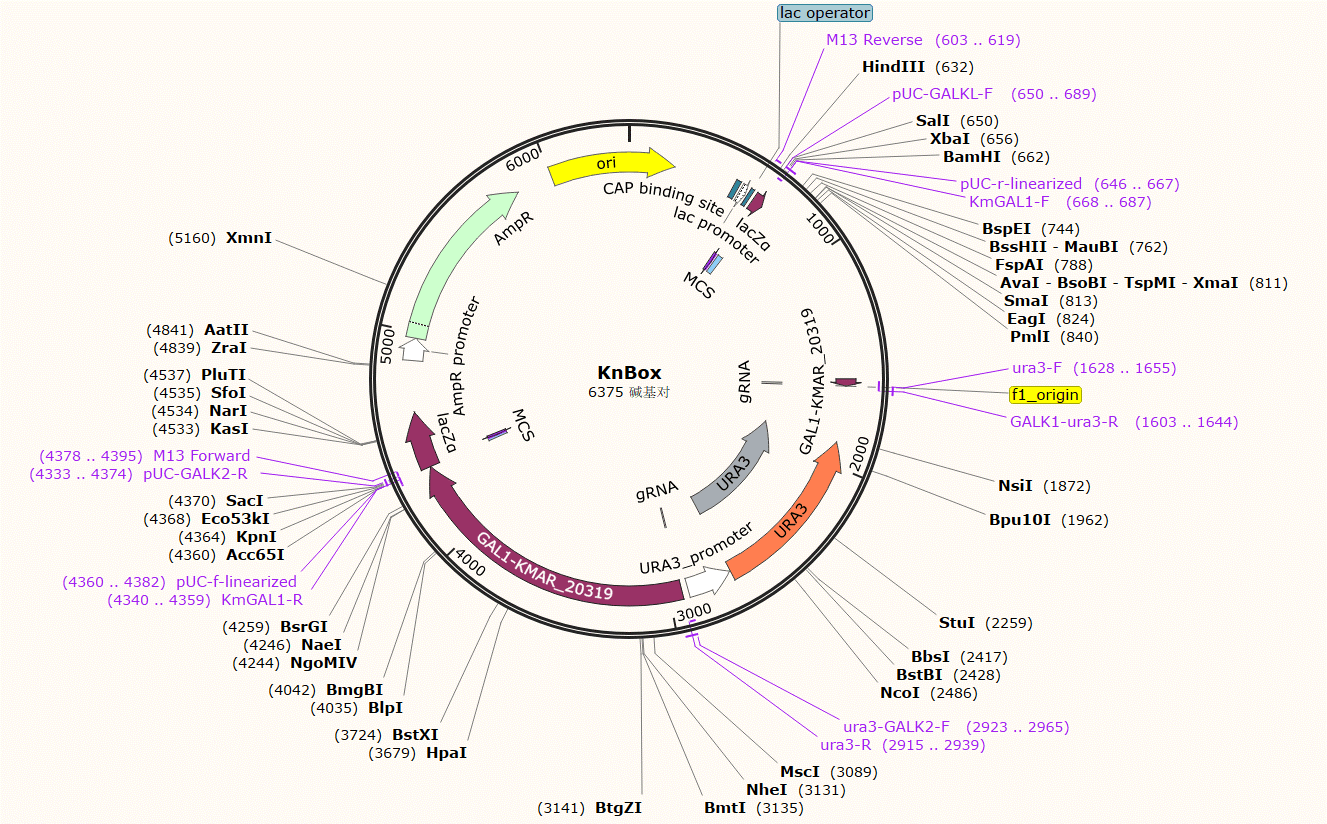
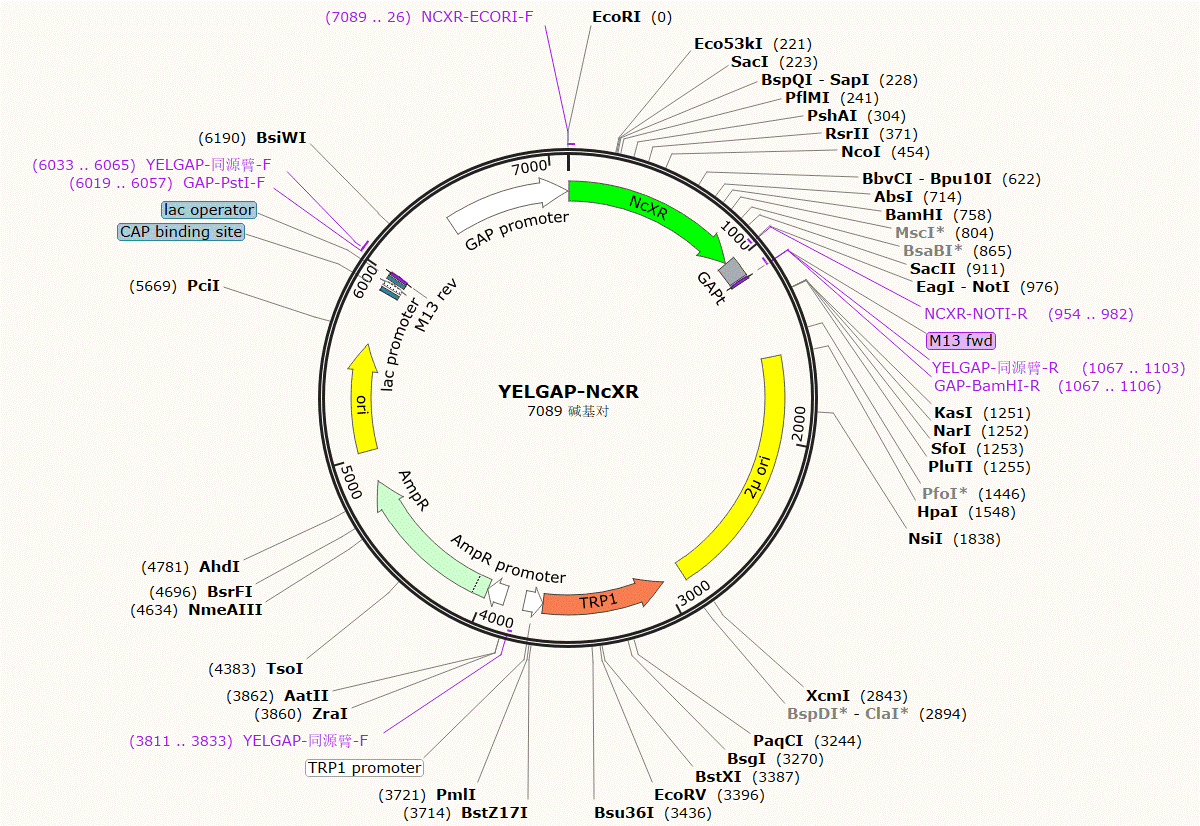
Transformation
We use the method of Lithium acetate to perform transformation
Reagents: YPD media (composed of 1% yeast extract , 2% tryptone and 2% glucose,extra 2% agar in solid) , 1ml Competent cell preparation buffer(800μl 50% PEG4000,100μl 1M DTT, 100μl 2M LiAc), SD solid media (depending on the labelled gene) , 10μl linear DNA fragments.
1.First,We streak-cultivate K.marxianus on YPD solid media and incubate overnight in a 37°C incubator ;
2.Use the inoculating needle to pick single colony from YPD plate and rinse the tip in 5ml YPD liquid media , cultivate overnight under the condition of 37°C and 230rpm;
3.Pipette 500μl suspension culture and transfer into 5ml YPD medium , cultivate under the condition of 37°C and 230rpm until the exponential growth phase (about 5 hours);
4.Centrifugate at 5000×g the culture for 1 minute , and then discard the supernatant . Add 500μl preparation buffer and use pipettes to blow and suck to resuspend yeast bodies;
5.Centrifugate at 5000×g the culture for 1 minute and discard the supernatant . Add 100μl preparation buffer and resuspend yeast bodies;
6.Add 10μl linear DNA fragments and 10μl Salmon sperm DNA(unnecessary) into the culture , vortex shake for 15 seconds ,and then 47°C heat shock for 15 minutes.
7.Spread appropriate amount of culture on a selective plates , and cultivate it in a 37°C incubator for 2~3 days.
Ferment
We use YPL and YPLG to ferment strains and detect content of tagatose by high performance liquid chromatography
Reagents:YPL media (consists of 1% yeast extract , 2% tryptone and 10% lactose) , YPLG (consists of 1% yeast extract , 2% tryptone , 8% lactose and 2% glycerol).
1.Cultivate strains in YPL (or YPLG) until OD600 reaches 5.0;
2.Pipette 1ml culture and transfer into 4ml YPL (or YPLG) in a test tube , cultivate it under the condition of 37°C and 230 rpm;
3.Every half a day sampling 0.8ml culture and store them at -4°C;
4.After 3 days separate and detect the content of tagatose by high performance liquid chromatography.
Sifting
Selective media
We choose Ura , Leu and Trp synthetic genes as markers to confirm whether each transformation is successful
Reagents:SD/-Ura , SD/-Leu , SD/-Trp media powder.
1.Prepare the media according to the instructions and add extra 2% agar in solid media.
2.Cultivate the strains on corresponding media according to the labelled gene.
If the transformation is successful , the yeast would self-synthesize the substance and grow on the plate.
Colony PCR
We do colony PCR to check again
Reagents:2 × Rapid Taq Master Mix , 20 mM NaOH , ddW.
1.Add 5-10μl NaOH in a PCR tube , and use the inoculating needle to pick single colony and rinse the tip in the PCR tube;
2.Heat the tube at 99.9°C for 30 minutes ensuring each cell is broken;
3.Add 2μl of each designed coupled primers , 25μl 2 × Rapid Taq Master Mix and ddW to final 50μl;
4.Carry out PCR under corresponding conditions;
5.Run an agarose gel electrophoresis and judge from the electrophoretogram.
Recombination
ClonExpress II One Step Cloning Kit C112(Vazyme biotechco.,ltd)was used for recombination
1.Calculation of the amount of vectors:
The optimal amount of vector for the recombination with ClonExpress II is 0.03 pmol, while the optimal amount of insert is 0.06 pmol (molar ratio of vector to insertion is 1:2), as roughly calculated as follows:
The optimal mass of vector = [0.02 x number of base pairs] ng (0.03 pmol)
he optimal mass of insert = [0.04 x number of base pairs] ng (0.06 pmol)
For example, when cloning an insert of 2 kb to a vector of 5 kb, the optimal mass of vector is 0.02 x 5000 = 100 ng, and the optimal mass of insert is 0.04 x 2000 = 80 ng. When using digested vectors and amplified inserts directly for recombination (without purification), the total volume of vectors and inserts should be ≤ 4 μL (1/5 of the total volume of recombination reaction system).
2.Calculate the amount of DNA for recombination by formula. Dilute linearized vectors and inserts before recombination to make sure the loading accuracy. The volume of each component loaded should be no less than 1 μL.
3.Set up the following reaction on ice:
| Components |
Recombination |
| Linearized Vectors |
X μL |
| Inserts |
Y μL |
| 5 x CE II Buffer |
4 μL |
| Exnase II |
2 μL |
| ddH2O |
to 20 μL |
a. X or Y indicates the amount of vector or insert calculated by formula
4.Gently pipette up and down for several times to mix thoroughly (DO NOT VOTEX!). Spin briefly to bring the sample to the bottom of the tube before reaction.
5.Incubate at 37 °C for 30 min and immediately place the tube at 4 °C or on ice.
▲It is recommended to use an instrument with high accurate temperature controlling system (i.e. a PCR instrument) for the reaction. The recombination efficiency can reach its peak at 30 min. Longer or shorter reaction time will decrease on the cloning efficiency.
▲The recombination product can be stored at -20℃ for one week. Thaw the product before transformation.
Plasmid extraction
RapidLyse Plasmid Mini Kit(Vazyme biotechco.,ltd)was used for plasmid extraction
1.Cell collection
Transfer 1-5 ml of overnight cultured bacteria into a 2ml centrifuge tube . Centrifuge at 12,000 rpm (13,800 × g) for 1 minute . Manage to aspirate out the supernatant.
2.Lysis
Add 600 μl of pre-cooled Buffer QLB into the centrifuge tube . Vortex shake immediately for 30 seconds and resuspend cells completely.
▲Complete resuspend to increase yield and avoid pollution of Genomic DNA.
3.Combination
Transfer the solution from the previous step into a adsorption column RapidLyse DNA Mini Columns . Centrifuge at 12,000 rpm (13,800 × g) for 30~60 seconds and discard the filtrate.
4.Rinse
Return the adsorption column to the collection tube , Add 600 μl Buffer QWB along the perimeter of the tube wall . Centrifuge at 12,000 rpm (13,800 × g) for 30~60 seconds and discard the filtrate.
5.Centrifuge again
Return the adsorption column to the collection tube and Centrifuge at 12,000 rpm (13,800 × g) for 30~60 seconds and discard the filtrate again.
▲Centrifuge again to completely get rid of the remaining liquid.
6.Elution
Place the adsorption column in a clean 1.5 ml centrifuge tube , add 50 ~ 100 μl of Buffer QEB to the center of the adsorption column membrane , centrifuge at 12,000 rpm (13,800 × g) for 30~60 seconds . Then store the plasmid DNA solution at -20℃ or use it in following experiments.
Electrophoresis
1.Prepared 1% or 0.7% agarose gel.
▲1% for 5kb marker ; 0.7% for 10kb marker.
2.Electrophoresis was performed at 120V for 20 min.
HPLC
| HPLC apparatus |
Recombination
-Column oven CTO-20A
-Solution transport unit LC-20ADXR
-HPLC autosampler SIL-20AXR
|
| Chromatographic column |
RezexTM ROA-Organic Acid H+ (8%) column (Phenomenex Inc., Torrance, CA) |
| Sample treatment |
Centrifuge at 14000rpm for 10min |
| Mobile phase |
0.0025mol / L H2SO4 |
| Column temperature |
75 °C / up to 90 °C |
| Flow rate |
0.3mL / min |
| Elution time |
10min |
| Pump pressure |
2.0 ~ 10.0MPa |