


Biological materials
| Names | Relevant characteristics | Source |
|---|---|---|
| E. coli DH5α | deoR endA1 gyrA96 hsdR17 (rk-mk+)recA1 relA1 supE44 thi-1 Δ(lacZYA-argF)U169 Φ80lacZ ΔM15F - λ - | Vazyme |
| E. coli BL21(DE3) | F-ompThsdS(rB-mB-) galdcm (DE3) | Vazyme |
| E. coli S2060 | F’ proA+B+ Δ(lacIZY) zzf::Tn10 lacIQ1 PN25-tetR luxCDE Ppsp(AR2) lacZ luxR Plux groESL / endA1 recA1 galE15 galK16 nupG rpsL ΔlacIZYA araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) proBA::pir116 araE201 ΔrpoZ Δflu ΔcsgABCDEFG ΔpgaC λ– | Addgene1 |
| E. coli S2208 | F’ proA+B+ Δ(lacIZY) zzf::Tn10 lacIQ1 PN25-tetR luxCDE Ppsp(AR2) lacZ luxR Plux groESL / endA1 recA1 galE15 galK16 nupG rpsL ΔlacIZYA araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) proBA::pir116 araE201 ΔrpoZ Δflu ΔcsgABCDEFG ΔpgaC λ– | Addgene |
Plasmids
| Names | Relevant characteristics | Purpose |
|---|---|---|
| pQE-80L | pBR322-KanR | Expression vector and negative control |
| pQE-80L-eGFP | pQE-80L-P22-T-PfdeAR-eGFP | positive control |
| pQE-80L-fdeR | pBR322-p22-fdeR-T-PfdeAR-eGFP | Biosensor |
| pQE-80L-p22-fdeR-Mutpre-A | pBR322-p22-fdeR-Mutpre-L268W | Site-directed mutagenesis |
| pQE-80L-p22-fdeR-Mutpre-B | pBR322-p22-fdeR-Mutpre-H170W | Site-directed mutagenesis |
| pQE-80L-p22-fdeR-Mutpre-C | pBR322-p22-fdeR-Mutpre-H170Y | Site-directed mutagenesis |
| pQE-80L-p22-fdeR-Mutpre-D | pBR322-p22-fdeR-Mutpre-H170W+L268W | Site-directed mutagenesis |
| pQE-80L-p22-fdeR-Mutpre-E | pBR322-p22-fdeR-Mutpre-H170Y+L268W | Site-directed mutagenesis |
| AP1 | pSC101Cm-T7-gⅢ-eGFP | positive selection |
| AP2-neg | pSC101Cm-T7-gⅢ-neg-RFP | positive selection |
| DP62 | A plasmid expresses mutagenic genes | Expression of mutagenic genes |
| pBT137-C2 | M13 carrier section with replica origin | Construction of selection phage |
| pBT137-D2 | M13 carrier fraction containing gⅢ | Construction of selection phage |
| AP-P-Mid2-T7RNAP | Contains Narigenin riboswitch Mid2 and T7RNAP | Construction of selection phage |
| AP-P-High2-T7RNAP | Contains Narigenin riboswitch High2 and T7RNAP | Construction of selection phage |
| pJC175e2 | Provides low copy of gⅢ | Construction of selection phage |
Primers
| No. | Name | Sequence (5’ -> 3’) | Function |
|---|---|---|---|
| 1 | H170Y-1-F | 5'-gcagacGTAccgttcgcggaagacctct-3' | Site-directed mutagenesis |
| 2 | SITE-1-R | 5'-cgcatatggtgcactctcagtaca-3' | Site-directed mutagenesis |
| 3 | H170Y-2-R | 5'-gaacggTACgtctgcgtggtctggcgc-3' | Site-directed mutagenesis |
| 4 | SITE-2-F | 5'-tgtactgagagtgcaccatatgcg-3' | Site-directed mutagenesis |
| 5 | L268W-1-F | 5'-ctcgccCCAcgacagcggactctccttgatc-3' | Site-directed mutagenesis |
| 6 | SITE-1-R | 5'-cgcatatggtgcactctcagtaca-3' | Site-directed mutagenesis |
| 7 | L268W-2-R | 5'-ctgtcgTGGggcgagatgcggcagatgat-3' | Site-directed mutagenesis |
| 8 | SITE-2-F | 5'-tgtactgagagtgcaccatatgcg-3' | Site-directed mutagenesis |
| 9 | H170W-1-F | 5'-gcagacCCAccgttcgcggaagacctct-3' | Site-directed mutagenesis |
| 10 | SITE-1-R | 5'-cgcatatggtgcactctcagtaca-3' | Site-directed mutagenesis |
| 11 | H170W-2-R | 5'-gaacggTGGgtctgcgtggtctggcgc-3' | Site-directed mutagenesis |
| 12 | SITE-2-F | 5'-tgtactgagagtgcaccatatgcg-3' | Site-directed mutagenesis |
| 13 | AP2-neg- GⅢneg-F | 5'-TTAAGACTCCTTATTACGCAGTATGTT-3' | Construction of AP2-neg |
| 14 | AP2-neg- GⅢneg-R | 5'-GGTGGCTCTTCCCAAATGGCTCAAGTCG-3' | Construction of AP2-neg |
| 15 | RFP-F | 5'-TGCTAGAACTGGCATGATTAAGCACCGGTGGAGTGAC-3' | Construction of AP2-neg |
| 16 | RFP-R | 5'-GTAATAAGGAGTCTTAAATGGCGAGTAGCGAAGACGT-3' | Construction of AP2-neg |
| 17 | AP2-neg-vector-F | 5'-TTTGGGAAGAGCCACCACCGGAACCG-3' | Construction of AP2-neg |
| 18 | AP2-neg- vector-R | 5'-TCATGCCAGTTCTAGCATAACCCC-3' | Construction of AP2-neg |
| 19 | AP2-pos-vector-F | 5'-CTCACCATTTAAGACTCCTTATTACGCAGTATGTTAG-3' | Construction of AP2-pos |
| 20 | AP2-pos-vector-R | 5'-ACAAGTAATCATGCCAGTTCTAGCATAACC-3' | Construction of AP2-pos |
| 21 | AP2-pos-eGFP-F | 5'-AGTCTTAAATGGTGAGCAAGGGCGAGGAG-3' | Construction of AP2-pos |
| 22 | AP2-pos-eGFP-R | 5'-TGGCATGATTACTTGTACAGCTCGTCCATGCC-3' | Construction of AP2-pos |
| 23 | pQE-pathway-vector-F | 5'-TAAGGCAGTTATTGGTGCCCTTA-3' | Construction of pQE-pathway |
| 24 | pQE-pathway-vector-R | 5'-TTCTCGAGGTGAAGACGAAAGG-3' | Construction of pQE-pathway |
| 25 | pQE-pathway-fragment-F | 5'-TCTTCACCTCGAGAATCAGCAGATGCCTGGCAGC-3' | Construction of pQE-pathway |
| 26 | pQE-pathway-fragment-R | 5'-CCAATAACTGCCTTAAAAAAATTACTTGTACAGCTCGTCCATGC-3' | Construction of pQE-pathway |
| 27 | pQE-SplitC-RBS-F | 5'-ACTAGACTACACTAAAAAGAGGACTATTCATCCGCTGCATGGCCCC-3' | Substitution of RBS of eGFP |
| 28 | pQE-SplitC-RBS-R | 5'-CGCATATGGTGCACTCTCAGTACAATCTG-3' | Substitution of RBS of eGFP |
| 29 | pQE-SplitD-RBS-F | 5'-TGTACTGAGAGTGCACCATATGCG-3' | Substitution of RBS of eGFP |
| 30 | pQE-SplitD-RBS-R | 5'-TCTTTTTAGTGTAGTCTAGTAATCCCGTTGATATTGATCAAATGGATTGTTTTG-3' | Substitution of RBS of eGFP |
| 31 | Mid2-random-F | 5'-CGATGCTGCTTATCGAAGTCTGA-3' | Nested PCR to obtain the fragment of Mid2 |
| 32 | Mid2-random-R | 5'-TGCAGAACGCGCCTGTTTATC-3' | Nested PCR to obtain the fragment of Mid2 |
| 33 | High2-random-F | 5'-AGGCGCGTTCTGCATTAGC-3' | Nested PCR to obtain the fragment of High2 |
| 34 | High-random-R | 5'-GCTGTAACAAGTTGTCTCAGGTGTTC-3' | Nested PCR to obtain the fragment of High2 |
| 35 | overlap-Mid2&High2-F | 5'-TACTACATAAATCAAACAGGGACGACGATGACACGAGA-3' | Overlap PCR to obtain Mid2-T7RNAP and High2-T7RNAP |
| 36 | overlap-Mid2-R | 5'-GTTAATCGTGTTCATAGATGCTCCTTCACTCGCATT-3' | Overlap PCR to obtain Mid2-T7RNAP and High2-T7RNAP |
| 37 | overlap-High2-R | 5'-GTTAATCGTGTTCATAGATGCTCCTTATTCCCCCC-3' | Overlap PCR to obtain Mid2-T7RNAP and High2-T7RNAP |
| 38 | overlap-T7RNAP-F | 5'-AGGAGCATCTATGAACACGATTAACATCGCTAAGA-3' | Overlap PCR to obtain Mid2-T7RNAP and High2-T7RNAP |
| 39 | overlap-T7RNAP-R | 5'-GGCATGAGCTCTTCAGCCTTACGCGAACGCGAAGTCC-3' | Overlap PCR to obtain Mid2-T7RNAP and High2-T7RNAP |
| 40 | overlap-AP-P-V-F | 5'-GTTCGCGTAAGGCTGAAGAGCTCATGCCAG-3' | Construction of the plasmid of AP-P-Mid2-T7RNAP and AP-P-High2-T7RNAP |
| 41 | overlap-AP-P-V-R | 5'-GTCGTCCCTGTTTGATTTATGTAGTAGACTAGAAGAGC-3' | Construction of the plasmid of AP-P-Mid2-T7RNAP and AP-P-High2-T7RNAP |
| 42 | ep-fdeR-F | 5'-ACCTCGAGAACGATGCTGCTTATCGAAGTCTGA-3' | Error-prone PCR to obtain fdeR |
| 43 | ep-fdeR-R | 5'-CTCGAATTCGGAGGAAACAAAG-3' | Error-prone PCR to obtain fdeR |
| 44 | ep-vec-F | 5'-TGTTTCCTCCGAATTCGAGGT-3' | Error-prone PCR to obtain pQE-80L used for vector |
| 45 | ep-vec-R | 5'-CATCGTTCTCGAGGTTCGTGATACGCCTATTTTTATAGGTTA-3' | Error-prone PCR to obtain pQE-80L used for vector |
Reagents
YEAST EXTRACT (OXOID, cat. no. LP0021) TRYPTONE (OXOID,cat. no. LP0042) NaCl (HUSHI,cat. no. 10019318)
X-Gal (Solarbio, cat. no. R0404)
Agar (Solarbio, cat. no. A8190)
Ampicillin (Solarbio, cat. no. A7094)
Streptomycin (Solarbio, cat. no. S8290)
Kanamycin (Solarbio, cat. no. K8020)
SapⅠ (New England BioLabs, cat. no. R0569S)
T4 DNA ligase (New England BioLabs, cat. no. M0202V).
IPTG (Isopropyl β-D- Thiogalactopyranoside ) (Solarbio, cat. no. I8070)
LB medium
FastPure Gel DNA (Nanjing Vazyme, cat. DC301-01)
FastPure Plasmid Mini kit (Nanjing Vazyme, cat. DC201-01)
DH5α Competent cell (Nanjing Vazyme, cat. C502-02)
ClonExpress MultiS One Step Cloning Kit (Nanjing Vazyme, cat. C113-01)
ClonExpress II One Step Cloning Kit (Nanjing Vazyme, cat. C112-01)
2×YT liquid media
(Add 2×YT media powder to a final concentration of 31 g/L in water. Mix to dissolve and then autoclave to sterilize at 121.0 °C. Store at room temperature indefinitely.)
2×YT agar
(Add 2×YT media powder and agar to a final concentration of 31 g/L and 1.5% (wtol), respectively, in water. Autoclave to sterilize at 121.0 °C. Store at room temperature in the dark for up to 6 months.)
2×YT top agar
(Melt 2×YT agar using a microwave until completely liquid. Dilute hot 2×YT agar with room temperature 2×YT liquid media to a final agar concentration of 0.5% or 0.6%(wtol) and mix thoroughly.)
LB liquid media
(Add YEAST EXTRACT, TRYPTONE, NaCl to a final concentration of 5 g/L,10 g/L,10 g/L in water, respectively.)
LB agar
(Add YEAST EXTRACT, TRYPTONE, NaCl, agar to a final concentration of 5 g/L, 10g/L, 10g/L, 1.5% (wtol), respectively, in water.)
1×PBS(Solarbio, cat. P1020)
10×PBS(Solarbio, cat. P1022)
Equipment
Biological shaker (Shanghai Zhichu Instruments, cat. no. ZHTY-50ES)
Benchtop centrifuge (Thermo Fisher Scientific, cat. no. 75007201)
Electrophoresis(Beijing Liuyi, cat. no. DYY-6C)
Absorbance Microplate Reader(TENCAN, cat. no.Tecan Infinite M200 NanoQuant)
Isolated thermostatic incubator (Shanghai SENXIN , cat. no. GNP-9080)
Gene Explorer(Bioer technology, cat. TC-96/G/H(b)B)
13 mm, 0.22-μm PVDF syringe filter(0.22 µm; Merck Millipore Ltd., cat. no. PR05538)
Gel imaging system(CLiN Qingxiang, cat. no. GenoSens2100)
Preparation
Extraciton of plasmid
According to the kit instructions for Nanjing Vazyme.
PCR fragment and vector
The obtained plasmids are subjected to PCR using different programs and primers.
PCR fragment and vector respectively with corresponding primers.
After agarose gel electrophoresis, we extract DNA from the gel. The products are stored at -20 ℃ for subsequent use.
Setting mutation conditions
Error-prone PCR
Use StarMut Random Mutagenesis Kit. Multiple small system (20μL) amplification pre-experiments were carried out, setting a series of StarMut Enhancer gradients. According to the PCR results and the mutation rate in the instructions, Enhancer volume (4μL) was determined.
Site-directed mutagenesis
With the help of two bioinformatic tools: AlphaFold2 and AutoDock 4.2.6, we spot three mutational residues: L268W、H170Y、H170W. Several pairs of degenerate primer were designed to randomly introduce mutations in fdeR.
Mutation
Error-prone PCR of fdeR
Using the error-prone PCR kit (Gene-star company) in a total volume of 25 μL using template 10 ng of pQE-80L-fdeR plasmid as template , 10μL 2×StarMut Random PCR Mix , 10 mM Gen 2-F, 10 μM Gen 2-R each, Star Mut enhancer 6 μL, sterile water was made up to 25 μL, 53.1℃) or error-prone PCR on fdeR fragments. Subsequently, the above fdeR gene fragments were sequenced by Sanger Sequence. Then based on the sequencing results, the number of mutated bases per thousand bases was calculated. Meanwhile, the linearized vector and the above fragments were assembled by ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China). E.coli BL21 was used for subsequent chemical transformation.
Site-Directed Mutation of FdeR
For construction of the single and double mutants of fdeR, the template the pQE-80L-fdeR plasmid was used along with a suitable combination of primers(Table S3).Three pairs of primers were designed to introduce three mutations (H170W,H170Y,L268W) on two sites in FdeR. The whole pQE-80L-fdeR plasmid was divided into two or three parts and then amplified by two or three pairs of primers(Table S3). The consequent two or three fragments with introduced mutations were combined with ClonExpress II One Step Cloning Kit or ClonExpress MultiS One Step Cloning Kit (Vazyme, Nanjing, China), which is a cloning kit based on homologous recombination. E.coli DH5α was used for subsequent chemical transformation. The plasmids from individual transformants were used to DNA sequence to confirm that correct mutation was obtained. Than, plasmids with correct mutations were transformed into E. coli BL21 . Randomly select individual colonies for fluorescence screening.
Bioinformatics analysis
Molecular Docking
We used AutoDock 4.2.6 28 for protein-small molecule docking. We obtained the 3D structure of the protein receptor using AlphaFold2 (Figure S10), and then performed a series of operations such as adding hydrogen, merging non-polar hydrogen, assigning Gasteiger charges, and storing it as a PDBQT format file. We obtained the planar conformation of liquiritigenin from PubChem, and then used OpenBabel to perform conformational conversion to form the optimal three-dimensional conformation required for docking and store it as a mol2 format file (Figure S9). Using AutoDockTools (ADT), we performed operations such as adding hydrogen and merging non-polar hydrogen atoms on liquiritigenin, and calculated the Gasteiger-Huiekel charges. The ligand was also stored as a PDBQT format file.
We chose the LGA algorithm for energy optimization, and set the relevant parameters of the Autogrid4 docking box and the Autodock4 docking program, and finally obtained a variety of docking conformations clustered by binding energy and RMSD - the computer selected the docking result with the lowest binding energy, and then used this docking conformation as a reference to compare other docking results. Compared with the control, those with a mean square deviation and structural difference of less than 0.5 Å were grouped into one group (cluster), and those with a difference of more than 0.5 Å were divided into another group. The different docking results in the same group were also sorted by binding energy and compared with the first one in the group. Those with a difference of less than 0.5 Å were left in the group, and those with a difference of more than 0.5 Å were re-grouped, and so on.
We used Discovery Studio to perform DNA 3D modeling of the fdeR gene promoter, and then used the protein as the receptor and the promoter as the ligand to perform DNA-protein docking on pyDockDNA. Then, according to the scoring ranking provided by pyDockDNA, we comprehensively judged and selected the docking results. The final screened conformation was analyzed and plotted using Pymol.
Prediction of single mutation sites
We use the software of Discovery Studio 2021 Client to predict the mutation sites. The receptor protein was applied CHARMm force field. Mutation stability was processed on single site using Calculate Mutation Energy (Binding) protocol of Discovery Studio 2021 Client.
Calculate Mutation Energy (Binding) protocol evaluates the effect of single-point mutations on protein stability. It performs amino-acid scanning mutagenesis on a set of selected amino-acid residues by mutating each of them to one or more specified amino-acid types. The mutation energy is the change in the protein’s stability caused by a mutation. It is measured by comparing how much energy the protein needs to fold in its original and mutated forms. The more energy the protein needs to fold, the less stable it is.
Molecular dynamics (MD) simulations
The molecular dynamics (MD) simulations were carried out by GROMACS 2020.3 software. The amber99sb-ildn force field and the general Amber force field (GAFF) were used to generate the parameter and topology of proteins and ligands, respectively. The simulation box size was optimized with the distance between each atom of the protein and the box greater than 1.0 nm. Then, fill the box with water molecules based on a density of 1. To make the simulation system electrically neutral, the water molecules were replaced with Cl- and Na+ ions. Following the steepest descent method, energy optimization of 5.0×104 steps was performed to minimize the energy consumption of the entire system, and finally to reduce the unreasonable contact or atom overlap in the entire system. After energy minimization, first-phase equilibration was performed with the NVT ensemble at 300 K for 100 ps to stabilize the temperature of the system. Second-phase equilibration was simulated with the NPT ensemble at 1 bar and 100 ps. The primary objective of the simulation is to optimize the interaction between the target protein and the solvent and ions so that the simulation system is fully pre-equilibrated. All MD simulations were performed for 50 ns under an isothermal and isostatic ensemble with a temperature of 300 K and a pressure of 1 atmosphere. The temperature and pressure were controlled by the V-rescale and Parrinello-Rahman methods, respectively, and the temperature and pressure coupling constants were 0.1 and 0.5 ps, respectively. Lennard-Jones function was used to calculate the Van der Waals force, and the nonbond truncation distance was set to 1.4 nm. The bond length of all atoms was constrained by the LINCS algorithm. The long-range electrostatic interaction was calculated by the Particle Mesh-Ewald method with the Fourier spacing 0.16 nm.
Transformation
Competent cells were thawed on ice. Pre-cooled plasmid mixture was added (each plasmid should < 100 ng; Each transformation of up to 3 plasmids), fully mixed after 30min on ice, 42℃ water bath, heat shock 90s(For S2060 and S2208 strains, heat shock 75s). Put back on ice and stand for 2-3min; Add 500 microliters of LB liquid medium, shake at 37℃, 200-300rpm/min, 1h; The bacterial solution was centrifuged at 5000rpm for 3min, the appropriate supernatant was removed, and then resuspended and coated in LB solid medium(For S2060 and S2208 strains, use 2×YT solid medium) containing appropriate antibiotics and 1.5% agar at 37℃ for 10-12h.
Screening
Fluorescence activated cell sorting (FACS) and 96-well plates screening
Cultures of E. coli BL21 with empty pQE-80L plasmids (Negative control) or transformed with plasmids containing eGFP reporter (Positive control). Library of FdeR mutants were grown overnight in 5 ml LB medium supplemented with 50 mg/mL kanamycin which grown on a biological shaker for 12 h at 220 rpm and 37℃. Cultures were diluted (1:100) into fresh media, grown until OD600~0.6 and induced with 0.5 mM liquiritigenin for 12 h.
The culture was diluted 10-fold in fresh liquid LB medium and analysed using FACS (BD FACSAria III, Institute of Soil Science, Chinese Academy of Sciences). A histogram plot was generated by sorting cells with empty pQE-80L plasmids and defining a negative gate as negative control. Meanwhile, using cells with plasmids containing eGFP reporter to designate a positive gate as a postive control. Two rounds of FACS counter-selection were constructed with liquiritigeni induction or naringenin induction.
In the 1st round, positive sorting was performed, collecting the population on the top 5%~10% of the histogram plot under the positive gate. At least 2.4×107 events were analysed by the flow cytometer, which can cover entire experimental libraries( 5.6×106 ). The collected cells were added into 5ml liquid LB medium supplemented with kanamycin to grow at biological shaker 220 rpm, 37℃, overnight. Cultures were diluted (1:100) into fresh LB medium, grown until OD600~0.6 and induced with 0.5 mM naringenin for 12 h as the samples of 2nd round.
In the 2nd round, negative sorting was performed, collecting the population on the bottom 5%~10% within the negative gate. At least 4.1×106 events were passed through the flow cytometer corresponding to 10-fold the initial library size. The sorted cells were plated on LB agar meium supplemented with 50 mg/mL Kanamycin overnight. Colonies were used for subsequent 96-well plates screening.
96 deep-well plates screening
The bacterial solution in 96 deep-well plate was centrifuged at 4000 rpm for 8 min after the induction culture completed, and then discard the supernatant. 1000 μL cold PBS buffer per well was pipetted and resuspended, and 200μl was taken into a 96-well fluorescent plate. Use a microplate reader to measure the fluorescence at Ex = 482 nm/Em = 524 nm and the absorbance at OD600.
Characterization and compensation test of biosensor
A concentration gradient of liquiritigenin ranging from 0 to 1 mM was introduced into 1 mL of bacterial culture containing kanamycin in a 96 deep-well plate, supplemented with 0.5 mM naringenin. The well-characterized variants were introduced into 5 mL of LB medium with kanamycin and incubated overnight at 37°C and 220 rpm on a shaker until reaching saturation. Plasmids were extracted using the FastPure Plasmid Mini Kit (Vazyme, Nanjing, China) and subsequently retransformed into BL21(DE3) competent cells (Vazyme, Nanjing, China). These cells were inoculated into 5 mL of kanamycin-supplemented LB medium and grown overnight on a Biological Shaker at 220 rpm and 37°C. Following cell resuspension, fluorescence intensity was measured using a microplate reader.
Characterization and compensation test of biosensor
When measuring fluorescence, we used 1×PBS to correct for background fluorescence and optical density of the PBS (FPPBS and ODPBS, respectively) for each imposed naringenin or liquiritigenin concentration. The normalized fluorescence for optical density was calculated as follows:
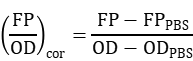
The resulting mean each concentration of naringenin or liquiritigenin, are expressed in arbitrary units (a.u.) and were fitted with the Hill function as follow using the weighted nonlinear least-squares algorithm (curve_fit, SciPy, Levenberg−Marquardt algorithm):
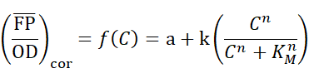
with C: the concentration of naringenin in the growth medium (mg/L); a: the basal normalized fluorescent signal (leaky expression, au); k: the relative maximum normalized fluorescent signal (au); n: the Hill coefficient (cooperativity, sigmoid character); KM: the Hill constant (half-maximal naringenin concentration, mg/L).
We apply a Noise parameter as an indicator for the overall mean error on the response of FdeR. The average relative error was calculated across the imposed naringenin or liquiritigenin concentrations, as follows:
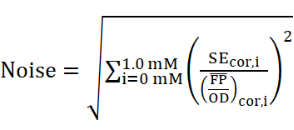
with SE cor,i the standard errors(SE) of the mean values for the imposed naringenin concentrations 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1 mM. We did not take the concentration 0.0 mM naringenin or liquiritigenin and the corresponding response signal into consideration, because they dominate the Noise parameter value.
DP6 mutation rate assay
DP6 was transformed into S2060 and plated on Chl-resistant 2×YT plates containing 100 mM D-Glucose. Monoclones were picked and shaken till saturation in 2×YT medium containing 25 mM D-Glu and Chl; the above bacterial solution was diluted 103-fold and grown to OD600 of 0.5-0.7. Samples were divided into two groups: one with 25 mM D-Glucose and the other with 25 mM arabinose, and incubated for 12h until saturation. Then,the bacterial solution was diluted by gradient and divided into two groups:one was applied to 2×YT plates containing 100 mM D-Glucose and Chl, the other was applied to 2×YT plates containing 100 mM D-Glucose and Rif. Culture them at 37°C for 18-24 h. The number of colonies of each culture on D-Glucose ± Rif plates was counted. Formula for calculating DP6 mutation rate (tbp: substituents per bp per generation): ubp = f/[R × ln(N//N0)], where f is the frequency of rifampin-resistant mutants (as compared with the glucose control), R is the number of unique sites yielding rifampin resistance (Liu et al. have identified only 21 sites across both rpoB clusters in their experiments),5 N is the final population size and N0 is the population size at which resistance is first observed (empirically determined to be B1.5107).
Transformation and storage of E. coli S2060 competent cells and strains prepared by chemical methods
Luciferase assay, phage propagation experiment and PACE were operated in the E. coli S2060 strain.
Competent cells are prepared by the method below. The saturated S2060 solution was added to a conical flask containing 2 x YT liquid medium (Solarbio) with appropriate antibiotics and diluted 100 times. The flask was then incubated in a shaker at 37℃ at 200-300 rpm/min until the OD600 of the bacterial solution was 0.4-0.6. We took and centrifuged 50 mL bacterial solution in the centrifugal tubes t 6500 rpm, 4℃ for 10 min. Discard the supernatant, and add 2 mL cold liquid LB and 2 mL cold 2×TSS (MgCl2, PEG 3350 and DMSO were added to LB medium at final concentrations of 20 mM, 10% and 5% respectively). Then, we froze the competent cells in the liquid nitrogen after mixing and dispensing them in the tubes and stored at -80℃.
As for transformation, 100 μL competent cells defrost on the ice; add pre-cooled plasmid mixture (each kind of plasmids should < 100 ng; 3 kinds of plasmids can be transferred at most) and 100 μL 1×KCM (KCl, CaCl2 and MgCl2 were added to LB medium at final concentrations of 100 mM, 30 mM and 50 mM respectively). Put it on the ice for 10 min after being mixedthoroughly, heat shock at 42 ℃ for 75 s; put it on the ice for 2-3 min; add 500 μL SOC solution in it and put it in the biological shaker at 200-300 rpm/min for 1 h; centrifuged the bacterial solution at 8000 rpm for 2 min, discard the supernatant; plate the bacterial solution on the 2×YT solid culture (1.5% agar) with proper antibiotics after the bacterial solution being resuspended; culture at 37 ℃ for 16-18 h. A saturated bacterial solution containing the appropriate antibiotic is mixed in equal proportions with sterile glycerol and stored frozen at -80 ℃.
Construction of selection phage
Golden Gate cloning was used to assemble selection phage (SP). For Golden Gate assembly, Sap Ⅰ (New England BioLabs) acted as IIS restriction endonuclease along with T4 DNA ligase (New England BioLabs). Golden Gate cloning reaction mixture was prepared as followes: 3-5 ng/kb/µL donor plasmids and 1-3 ng/kb/µL receptor (pBT137-SplitC and pBT137-SplitD). And the mixtures experienced thermal cycles (37 ℃ for 5 min,16 ℃ for 5 min, 40 cycles, 60 ℃ 5 min, 4 ℃). After this process, assemblies were transferred into S2208 competent cells.
Activity-independent phage plaque assay
Saturated S2208 bacterial broth was diluted with 2×YT liquid medium containing Amp and shaken at 37°C to OD600 : 0.6-0.9. Phage was diluted 3 times with sterilized water in a gradient of 102 . Transfer 10 μL of each different concentration of phage to a centrifuge tube. Add 150 μl of diluted S2208, then 20 μL of X-Gal (dissolve X-Gal in DMF to a final concentration of 2% (wt/vol)) solution, leave for 5 min, add 1mL of hot 2×YT top agar, mix well and pour the above mixture onto 2×YT solid agar tetrads at 37 °C for 5-8 h. Calculate the phage titer: Phage titer number of phage spots in the corresponding quadrant × dilution × 100.
Isolation of colonies
Gently touch the plaque with the tip of a P10 pipette. Place the pipette tip into 2-3 mL of DRM3, grow for 16-20 h in a biological shaker at 37℃. Centrifuge at 8000 g for 2 min. The supernatant was collected and filtered in a 1 ml syringe equipped with a PVDF filter (13 mm 0.22 μm) to obtain a phage stock solution. Its concentration was determined using an activity-independent phage plaque assay. POI fragments can also be amplified by PCR for sequencing.
Activity-dependent plaque test
S2060 saturated bacterial solution transformed with AP was diluted 1000 times into DRM3 medium containing appropriate antibiotics, and the culture was shaken at 37 ℃ until OD600 grow to 0.4-0.6. Cells were infected with phage at a starting titer of approximately 104 PFU/ml and incubated at 37 ℃ for 16-20 h on a biological shaker. Centrifuge at 8000 g for 3 min. The supernatant was collected and filtered in a 1ml syringe equipped with a PVDF filter (13 mm 0.22 μm) to obtain phage stock solution, which was stored at -4 ℃.
Purification of SP
In the above transformation step, the above plasmids were replaced with a mixture product of assembled SP plasmids, heat shocked for 75 s, left on ice for 2-3 min, and then the cell mixture was directly diluted into 10ml of antibiotic-free 2×YT liquid medium at 37 ℃ for 16-18 h at 200-300 rpm/min on a biologicalshaker. 2 mL of bacterial solution was put into a centrifuge tube and centrifuged at 8000 g for 3 min. The supernatant was collected and filtered in a 1 ml syringe (Nanjing Dingbei Biotechnology co., LTD.) equipped with a PVDF filter (13 mm 0.22 μm) (Nanjing Dingbei Biotechnology co., LTD.) to obtain the phage stock solution and stored at -4℃.
1. B.P. Hubbard, A.H. Badran J.A. Zuris et al, Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat Methods 12, 939-42, (2015).
DOI: http://doi.org/10.1038/nmeth.3515
2. A.H. Badran, V.M. Guzov Q. Huai et al, Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58-63, (2016).
DOI: http://doi.org/10.1038/nature17938
3. J.C. Carlson, A.H. Badran D.A. Guggiana-Nilo et al, Negative selection and stringency modulation in phage-assisted continuous evolution. Nat Chem Biol10, 216-22, (2014).
DOI: http://doi.org/10.1038/nchembio.1453
4. G.M. Morris, R. Huey W. Lindstrom et al, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 30, 2785-91, (2009).
DOI:http://doi.org/10.1002/jcc.21256
5. A.H. Badran D.R. Liu, Development of potent in vivo mutagenesis plasmids with broad mutational spectra.Nat Commun, 6 8425, (2015).
DOI: http://doi.org/10.1038/ncomms9425






