To investigate the effects of site-directed mutagenesis, the wild-type (WT) and three mutant enzymes (Design1, Design2_IM(The intermediate construct from Design 2) and Design2) were expressed and purified. Quantification of the purified proteins revealed that the mutations significantly impacted expression levels. The WT enzyme exhibited a robust expression yield of 3.27 mg/L. In stark contrast, all three mutants showed a substantial reduction in yield. The final yields for Design2_IM, Design1, and Design2 were 0.788mg/L, 1.32 mg/L, and 1.08 mg/L, respectively. These levels correspond to approximately 24%, 40%, and 33% of the WT expression level.
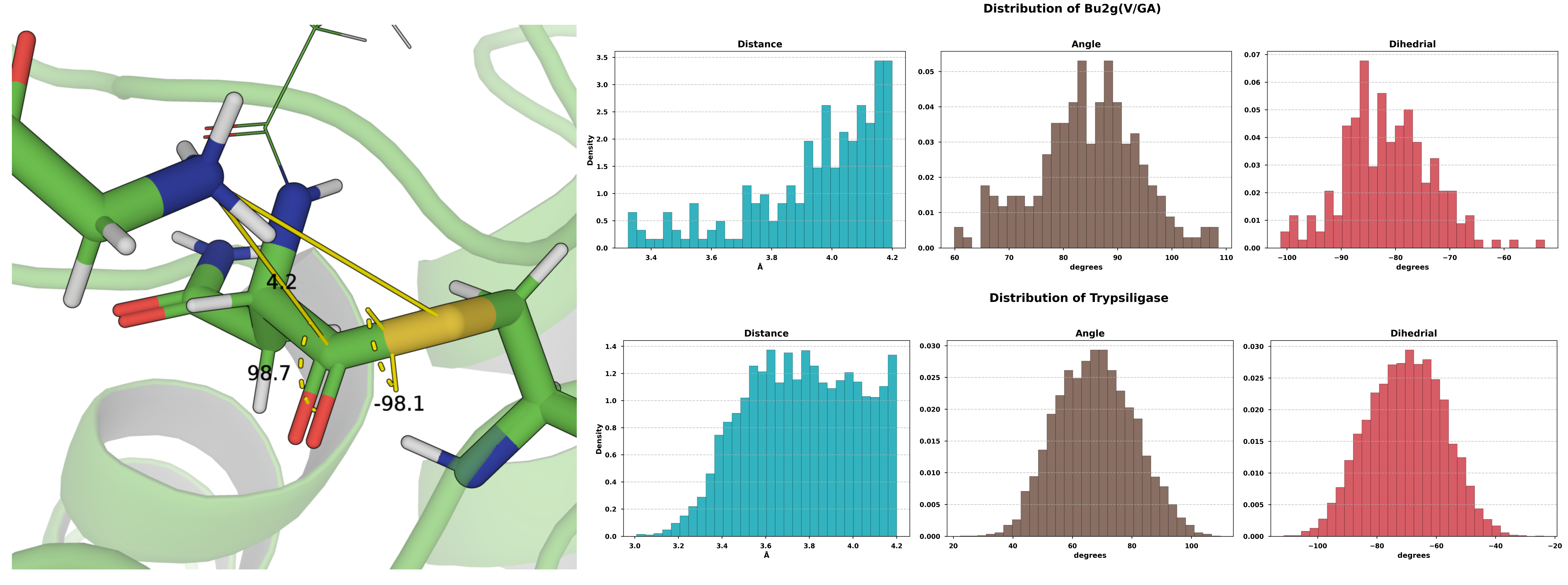
Figure 3. Characterization of the two ligation systems within a 4 Å proximity.
To elucidate the key determinants of the covalent modifications on reactivity, the hydrolytic and ligation activities of several enzyme systems were evaluated (summarized in Table 2). Herein, the acyl donor refers to the portion of the hydrolysis substrate that remains covalently attached to the catalytic residue following the formation of the ester intermediate, whereas the acyl acceptor is the ligation substrate. Figure 4 shows the distribution of the attack angle between water molecules and the ester intermediate across the five simulated hydrolytic reaction systems. Perhaps due to the significant deviation of the initial structure from the native binding pose, the simulated ligation systems of Bu2g(V/GA) and Trypsiligase exhibited mutual approximation between the N-terminus of the acyl-acceptor and the electrophilic center of the acyl-donor. Furthermore, the resulting attack angles and attack dihedrals exhibited a defined distribution pattern.
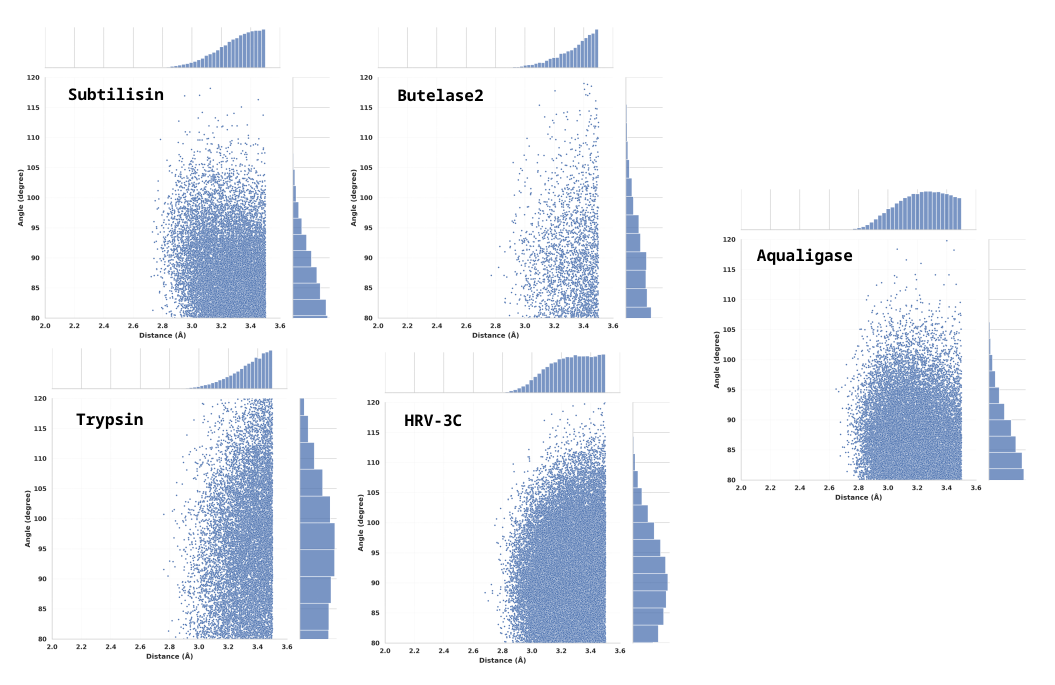
Figure 4. Statistical Distribution of the local Water-Oxygen Attack Angle within 3.5 Å of the Ester Intermediate for Four Wild-Type Hydrolases (Subtilisin,Butelase2,Trypsin,HRV-3C) and the Aqualigase Variant with a Water-Shielding Pocket Design.
To evaluate the catalytic activity of the wild-type (WT) and mutant enzymes (19, 105, 143), we
sought to measure their performance in a transpeptidation (ligation) reaction. Reactions were
conducted in the presence of potential nucleophiles: a peptide (32GITRR, at a 32:1 molar ratio to the
substrate) and an amino acid (1000Gly, at a 1000:1 molar ratio). Notably, HPLC analysis revealed no
detectable formation of the expected ligation product in any of the tested conditions. The only
observed product corresponded to the hydrolytic cleavage of the substrate. Consequently, our
analysis focused on the hydrolytic activity of the enzymes under these different conditions,
quantified as the ratio of the hydrolytic product peak area to the remaining substrate peak area. The
results after 12 and 24 hours of reaction are summarized in Table 1.
| Enzyme | Condition | Hydrolysis Ratio (12h) | Hydrolysis Ratio (24h) |
|---|---|---|---|
| WT | Pure Hydrolysis | 4.882871 | 5.870103 |
| 32GITRR | 6.287923 | 7.013444 | |
| 1000Gly | 9.710145 | 8.505435 | |
| Design1 | Pure Hydrolysis | 4.831418 | 6.653915 |
| 32GITRR | 4.254176 | 6.278571 | |
| 1000Gly | 7.637177 | 8.229491 | |
| Design2_IM | Pure Hydrolysis | 6.467882 | 7.574692 |
| 32GITRR | 5.53022 | 7.127273 | |
| 1000Gly | 5.215517 | 7.909449 | |
| Design2 | Pure Hydrolysis | 5.21548 | 12.1319 |
| 32GITRR | 4.037259 | 7.263775 | |
| 1000Gly | 6.441385 | 8.73125 |
Table 1. Hydrolytic activity of WT and mutant enzymes in the presence of different nucleophiles. Values represent the ratio of the peak area of the hydrolysis product to the peak area of the remaining substrate, as determined by HPLC. “Pure Hydrolysis” serves as the control. “32GITRR” and “1000Gly” represent reactions supplemented with the GITRR peptide (peptide:substrate ratio of 32:1) and glycine (glycine:substrate ratio of 1000:1), respectively. All reactions were conducted under identical conditions.
In our pursuit of engineering HRV-3C wild-type (WT) and its variants into efficient peptide ligases, we assayed their catalytic activities in the presence of potential external nucleophiles—the GITRR peptide and glycine. Contrary to our design objective, the desired ligation product was not detected under the tested aqueous conditions, with its concentration remaining consistently below the limit of detection of the HPLC method. Instead, the predominant catalytic activity observed was substrate hydrolysis. Consequently, our investigation pivoted to a systematic characterization of how these additives influence the intrinsic hydrolytic function of the enzymes. The activity, quantified by the peak area ratio of the hydrolysis product to the remaining substrate, is presented in Table 1. In the control (pure hydrolysis), mutant Design2 was identified as the most hydrolytically active variant, exhibiting a 24-hour activity ratio (12.13) markedly higher than that of the WT (5.87) and other mutants. Intriguingly, rather than facilitating ligation, these nucleophilic compounds served as modulators of hydrolytic activity in an enzyme-specific manner. For the highly active mutant Design2, both 32GITRR and 1000Gly exerted an inhibitory effect, reducing its activity ratio to 7.26 and 8.73, respectively. In sharp contrast, the WT enzyme was consistently activated by these additives, with its 24-hour activity ratio increasing from 5.87 to 7.01 and 8.50 in the presence of 32GITRR and 1000Gly, respectively. These findings suggest that despite their inefficacy as ligation substrates, these molecules can interact with the enzyme and allosterically modulate its canonical hydrolytic pathway.
The strategy of rationally designing the HRV-3C protease to construct its hydrophobic pocket for developing novel ligases stems from a thorough evaluation of existing mainstream ligase technologies. First, PAL family ligases derived from asparaginyl endopeptidase (represented by Butelase 1) face bottlenecks in expression and purification. Despite their high catalytic efficiency and short recognition sequence (the tripeptide motif D/N-HV), these enzymes originate from the plant butterfly pea, making recombinant expression challenging[4]. The complex activation process required for their proenzyme form is difficult to precisely control, affecting experimental reproducibility. While the short recognition sequence facilitates handling, it increases the risk of non-specific reactions. Practical implementation also requires high substrate concentrations to drive the reaction, potentially triggering substrate inhibition and escalating costs. Additionally, Subtiligase derived from Bacillus subtilis protease faces constraint: substrate pretreatment requirements.[27] While this enzyme demonstrated the feasibility of serine protease-catalyzed ligation, it requires preactivation of the substrate C-terminus into a high-energy ester. This additional step complicates operations, increases costs, and may induce side reactions like racemization, limiting its application in peptide modification. Finally, Staphylococcus aureus Sortase A suffers from core defects: thermodynamic reversibility and low kinetic efficiency. Its reversible catalytic mechanism limits conversion rates, necessitating excessive use of donor substrates, which increases costs and purification complexity. The reaction exhibits sensitivity to conditions and poor reproducibility, making standardized workflow establishment challenging[28]. Slow catalytic rates often necessitate high substrate concentrations, compromising economic viability. Its application in cyclic peptide synthesis is particularly limited due to the generation of lengthy “scar” sequences and the requirement for substrates exceeding 19 amino acids to avoid oligomerization side reactions. In summary, existing ligases exhibit significant shortcomings in expression complexity, substrate pretreatment requirements, and reaction reversibility. Consequently, there is an urgent need to develop novel ligases that can be efficiently expressed in E. coli, require no preactivation, directly utilize native carboxylate substrates, and exhibit irreversible reactions. The HRV-3C protease offers advantages such as high prokaryotic expression yield, excellent stability, and strong specificity (recognizing the long sequence LEVLFQ-G), making it an ideal starting point for engineering. Through rational design to construct a hydrophobic pocket, its binding capacity with hydrophobic substrates can be enhanced, optimizing the reaction pathway and thereby achieving a functional transformation from a hydrolase to an efficient ligase.
| Reaction | System | Environment | Acyl donor | Acyl acceptor |
|---|---|---|---|---|
| Hydrolytic | Subtilisin | pH8, 25°C | Suc-AAPF | --- |
| Aqualigase | pH8, 40°C | Suc-AAPF | --- | |
| HRV-3C | pH8, 4°C | LEVLFQ | --- | |
| Butelse2 | pH7, 37°C | ISYRN | --- | |
| Trypsin | pH8, 37°C | GGGY | --- | |
| Ligation | Aqualigase | pH8, 40°C | Suc-AAPF | AF-NH2 |
| Subtiligase | pH8, 25°C | Suc-AAPF | AF-NH2 | |
| HRV-3C | pH8, 4°C | LEVLFQ | GITRR | |
| Bu2g(V/GA) | pH7, 37°C | GISTKSIPPISYRN | GISTKSIPPISYRN | |
| Trypsiligase | pH8, 37°C | GGGY | RNGGG |
Predicated on the assumption that a portion of the mutant proteins retained correct folding, the lack of observed ligation activity indicates that the strategy of constructing a “water-proof pocket” solely through the introduction of hydrophobic residues is inadequate to surmount the potent hydrolytic background. Water acts as a ubiquitous and highly competitive nucleophile in aqueous environments, with an effective concentration reaching approximately 55 M. Furthermore, the active sites of natural hydrolases have undergone extensive evolutionary optimization to exploit this abundance efficiently. While our cMD simulations revealed that the distribution of water attack angles in the mutants deviated from that of the WT, this alteration was likely insufficient to fundamentally preclude the hydrolytic reaction. Consequently, dismantling this evolutionarily refined “hydrolysis machinery” may necessitate structural remodeling that is more drastic or intricate than previously envisioned.
This insight pivots our strategic focus for future designs. Our initial design of the hydrophobic pocket prioritized the direct exclusion of water; however, future work should specifically target the stage of competition with hydrolysis: the attack by the peptide nucleophile substrate. Characterizing the kinetics of this peptide’s attack will provide a direct and far more meaningful metric for assessing our progress in creating a truly competitive ligation pathway.
[12] M. Tiberti et al., “MutateX: an automated pipeline for in silico saturation mutagenesis of
protein structures and structural ensembles,” Briefings in bioinformatics, vol. 23, no. 3, p.
bbac74, 2022.
[13] J. Abramson et al., “Accurate structure prediction of biomolecular interactions with AlphaFold
3,” Nature, vol. 630, no. 8016, pp. 493–500, 2024.
[14] O. Trott and A. J. Olson, “AutoDock Vina: Improving the speed and accuracy of docking with a
new scoring function, efficient optimization, and multithreading,” Journal of Computational
Chemistry, vol. 31, no. 2, pp. 455–461, 2010, doi: https://doi.org/10.1002/jcc.21334.
[15] T. Lu, “A comprehensive electron wavefunction analysis toolbox for chemists, Multiwfn,” The
Journal of Chemical Physics, vol. 161, no. 8, p. 82503, 2024, doi: 10.1063/5.0216272.
[16] T. Lu, “Tian Lu, Sobtop, Version 1.0 (dev 5) http://sobereva.com/soft/Sobtop (accessed on 2
10,2025),” vol. 0, p. , 2025.
[17] M. J. Abraham et al., “GROMACS: High performance molecular simulations through multilevel
parallelism from laptops to supercomputers,” SoftwareX, pp. 19–25, 2015, doi: https://doi.
org/10.1016/j.softx.2015.06.001.
[18] C. Tian et al., “ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against
Quantum Mechanics Energy Surfaces in Solution,” Journal of Chemical Theory and
Computation, vol. 16, no. 1, pp. 528–552, 2020, doi: 10.1021/acs.jctc.9b00591.
[19] S. Izadi, R. Anandakrishnan, and A. V. Onufriev, “Building Water Models: A Different
Approach,” The Journal of Physical Chemistry Letters, vol. 5, no. 21, pp. 3863–3871, 2014, doi:
10.1021/jz501780a.
[20] D. A. Case et al., “AmberTools,” Journal of Chemical Information and Modeling, vol. 63, no. 20,
pp. 6183–6191, 2023, doi: 10.1021/acs.jcim.3c01153.
[21] J. Wang and Y. Miao, “Ligand Gaussian Accelerated Molecular Dynamics 3 (LiGaMD3):
Improved Calculations of Binding Thermodynamics and Kinetics of Both Small Molecules and
Flexible Peptides,” Journal of Chemical Theory and Computation, vol. 20, no. 14, pp. 5829–5841,
2024, doi: 10.1021/acs.jctc.4c00502.
[22] Z. Zhang, W. X. Shen, Q. Liu, and M. Zitnik, “Efficient generation of protein pockets with
PocketGen,” Nature Machine Intelligence, pp. 1–14, 2024.
[23] Z. Lin et al., “Evolutionary-scale prediction of atomic-level protein structure with a language
model,” Science, vol. 379, no. 6637, pp. 1123–1130, 2023, doi: 10.1126/science.ade2574.
[24] M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente, and E. Moreno, “gmx_MMPBSA:
A New Tool to Perform End-State Free Energy Calculations with GROMACS,” Journal of
Chemical Theory and Computation, vol. 17, no. 10, pp. 6281–6291, 2021, doi: 10.1021/
acs.jctc.1c00645.
[25] J. L. Watson et al., “De novo design of protein structure and function with RFdiffusion,” Nature,
vol. 620, no. 7976, pp. 1089–1100, 2023.
[26] J. Dauparas et al., “Atomic context-conditioned protein sequence design using LigandMPNN,”
Biorxiv, pp. 2023–2012, 2023.
[27] T. Nuijens, A. Toplak, M. Schmidt, A. Ricci, and W. Cabri, “Natural occurring and engineered
enzymes for peptide ligation and cyclization,” Frontiers in Chemistry, vol. 7, p. 829, 2019.
[28] R. Warden-Rothman, I. Caturegli, V. Popik, and A. Tsourkas, “Sortase-Tag Expressed Protein
Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step,”
Analytical Chemistry, vol. 85, no. 22, pp. 11090–11097, 2013, doi: 10.1021/ac402871k.

This
study aimed to convert the HRV-3C protease, a highly specific cysteine hydrolase, into a novel
peptide ligase through rational design. Our primary strategy involved computationally engineering
a hydrophobic active-site pocket to suppress the enzyme’s intrinsic hydrolytic activity by sterically
hindering the access of nucleophilic water molecules.