The pET-47b(+)-NT-HRV3CP plasmid was used as the template for site-directed mutagenesis. Primers containing the desired mutations were synthesized by Tsingke Biotechnology Co., Ltd. (Beijing, China). Point mutations were introduced using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme, Cat: C214-02) according to the manufacturer’s protocol. The mutagenesis reaction products were then transformed into E. coli DH5α chemically competent cells (Sangon Biotech, Shanghai, China). Transformants were selected on Luria-Bertani (LB) agar plates supplemented with 50 μg/mL kanamycin. Positive colonies were cultured, and the mutant plasmids were extracted using the FastPure Plasmid Mini Kit (Vazyme, Cat: DC201-01). The primers used to construct Mutant 1 and Mutant 2 are listed in Supplementary Table S1. The successful introduction of all mutations was verified by Sanger sequencing, performed by Tsingke Biotechnology Co., Ltd. (Beijing, China).
The NT*-HRV3CP[11] protein was expressed in E. coli BL21(DE3) cells (Sangon Biotech, Shanghai,
China). To prepare the protein, the pET-47b(+) plasmid was transformed into E. coli BL21(DE3) cells,
which were then cultured at 37 °C in LB medium containing 50 μg/mL kanamycin. An overnight
culture of 2.5 mL was used to inoculate 250 mL of TB medium supplemented with 50 μg/mL
kanamycin. The culture was grown at 37 °C until the optical density at 600 nm (OD600) reached 0.6–
1. Subsequently, the temperature was lowered to 18 °C, and protein expression was induced with 0.5
mM isopropyl-β-D-thiogalactopyranoside (IPTG) for 16 hours.
After expression, the cells were harvested by centrifugation and resuspended in Buffer A (50
mM Tris-HCl pH 7.5, 300 mM NaCl, 5% glycerol, 10 mM imidazole) supplemented with 0.1% Triton
X-100, followed by lysis using a sonicator. The cell lysate was clarified by centrifugation at 12,000
rpm for 1 hour. The resulting supernatant was loaded onto a 1 mL HisTrap FF affinity
chromatography column connected to an ÄKTA start chromatography system (Cytiva, USA). The
column was washed with 20 column volumes of Buffer B (identical to Buffer A, but with 20 mM
imidazole) and the protein was then eluted with 3 column volumes of Buffer C (identical to Buffer A,
but with 500 mM imidazole). The eluted protein was buffer-exchanged into buffer D (50 mM Tris-
HCl pH 7.5, 300 mM NaCl, 1 mM DTT) and concentrated using an Amicon ultrafiltration centrifugal
tube (Merck Millipore, USA) with a molecular weight cut-off of 10 kDa. Afterwards, the protein was
stored in 10% glycerol at −80 °C after snap freezing in liquid nitrogen. Protein concentration was
measured with a Unano-1000 Micro-Spectrophotometer (Hangzhou UMI Instrument Co., Ltd.,
Hangzhou, China) using the 280 nm extinction coefficient of 11,460 M⁻¹cm⁻¹ calculated for NT*-
HRV3CP.
The enzymatic activity of NT*-HRV3CP and its variants was evaluated in a 100 μL reaction system. For the hydrolysis reaction, the mixture contained 20 mM of the peptide substrate LEVLFQAPG and 0.2 mM of the enzyme, dissolved in Buffer D. For the ligation reaction, the mixture was additionally supplemented with 640 mM of the nucleophilic peptide GITRR. All reactions were incubated at 25 °C for 24 hours and subsequently stopped by the addition of 0.1% formic acid. The quenched reactions were then centrifuged at 12,000 rpm for 30 minutes, and 80 μL of the supernatant was transferred to an HPLC vial for analysis.
Reaction products were analyzed by high-performance liquid chromatography (HPLC) on a C8 column. The injection volume for each run was 20 μL. The mobile phase consisted of solvent A (water with 0.1% trifluoroacetic acid) and solvent B (acetonitrile with 0.1% trifluoroacetic acid). Separation was performed at a flow rate of 1.0 mL/min using the following gradient: a linear increase from 2% to 95% solvent B over 35 minutes, followed by a 4-minute wash with 95% solvent B, and finally, a 3-minute equilibration with 2% solvent B. The elution signal was monitored by absorbance at 220 nm.
The HRV-3C wild-type protein structure was obtained from the Protein Data Bank (PDB ID: 2B0F). We used all these structures to estimate the effects of amino acid substitutions on protein stability using FoldX. The RepairPDB function was run 5 times to repair incorrect torsion angles, VanderWaals clashes and total energy of the structure. We then used the MutateX[12] pipeline to perform an in silico deep mutational scanning of the whole protein, running the 3 rounds BuildModel function from FoldX and computing the average difference in Gibbs free energy between the mutant and the wild type.
All protein monomers and protein-hydrolytic substrate complex structures were modeled using AlphaFold3[13]. The protonation states of amino acid residues were determined using the H++ webserver. Prior to the production simulations, a 20 ns stability validation was conducted. Molecular docking was performed with Autodock Vina[14] to obtain an ester intermediate-linked substrate complex. The topology and force field parameters for the enzyme-substrate ester intermediate structures were described via additional parametrization, which modeled the deprotonated catalytic residue (SER or CYS) forming an ester intermediate with the hydrolytic substrate.[15], [16]
Conventional molecular dynamics (cMD) simulations were conducted using the GROMACS 2025.2[17] package. Unless otherwise specified, the Amber ff19SB[18] force field along with the OPC[19] water model were employed for all simulations. All system were neutralized with an appropriate number of ions. On top of neutralization, additional K+ and Cl– ions were added to bring the final ion concentration to 150 mM. Periodic boundary conditions and dispersion corrections for energy and pressure were applied throughout all the minimization and cMD simulations.A nonbonded interaction cutoff of 10 Å was implemented, with long range electrostatic interactions treated via the Particle Mesh Ewald method.
First, the system was minimized using the steep method for 100000 steps,ensuring that the maximum force was reduced to 100 kJ·mol¹·nm¹. No restraints were applied in minimization. After the mininimization, 2ns NPT simulation was carried out at target temperature and 1 atm to further equilibrate the system and correct the density. The temperature coupling was controlled using the Vrescale method (τₚ = 0.5), and pressure coupling was initially managed using the C-rescale method (τₚ = 2.0). A 1-fs timestep was used. No restraints were applied. During the equilibration phase, subsequent NPT equilibration employed pressure coupling via the C-rescale barostat with isotropic scaling (τₚ = 2.0). Temperature coupling was maintained using the V-rescale algorithm (τₜ = 0.2).
Then each system underwent NPT equilibration for a 20 ns duration. All bonds involving hydrogen atoms were constrained with the LINCS algorithm, enabling a 2 fs integration time step. Finally, three independent replicates of 100 ns simluations were carried out for production and this trajectory was used for data analysis.
The force field parameters for the non-standard residue and additional bonded parameters derived from the covalently-linked ester intermediate were obtained from the GROMACS database. Detailed parameters for all atoms, bonds, angles, and dihedrals are provided in the Supporting Information.
The AMBER24[20] package was used to preform LiGaMD3[21]. LiGaMD3 (igamd = 28) was performed to sample the interaction from ester intermediate-linked substrate complex. Simulations utilized the Amber ff19SB force field and OPC3 water model. Temperature was maintained at their respective physiological temperatures using the Langevin thermostat (ntt = 3, and gamma_ln = 5.0). Pressure was maintained isotropically (ntp = 1, and taup = 2.0). Nonbonded interactions were handled with a cutoff of 10.0 Å (cut = 10.0).
In LiGaMD3, three distinct boosts are applied: one on the non-bonded interaction energy of the substrate, the second one on the remaining non-bonded potential energy of the system, and a third one on the system bonded potential energy. For upper limit of the standard deviation of the potential boost, σP were set to 3.0 kcal/mol (The Bu2g(V/GA) system is 4.5 kcal/mol). Default values were used for other input parameters. GaMD consisted of the pre-equilibration stage, equilibration stage and production stage. The previously equilibrated snapshot prepared from the unbiased conventional MD was used as the initial configuration of GaMD. The pre-equilibrated system was subjected to additional unbiased cMD of 6 ns as pre-equilibration before collection of potential data for 20 ns for the determination of boost potential. Three independent replicas of 100 ns GaMD simulations were done for the production stage using fixed boost statistics and coordinates being saved every 2 ps.
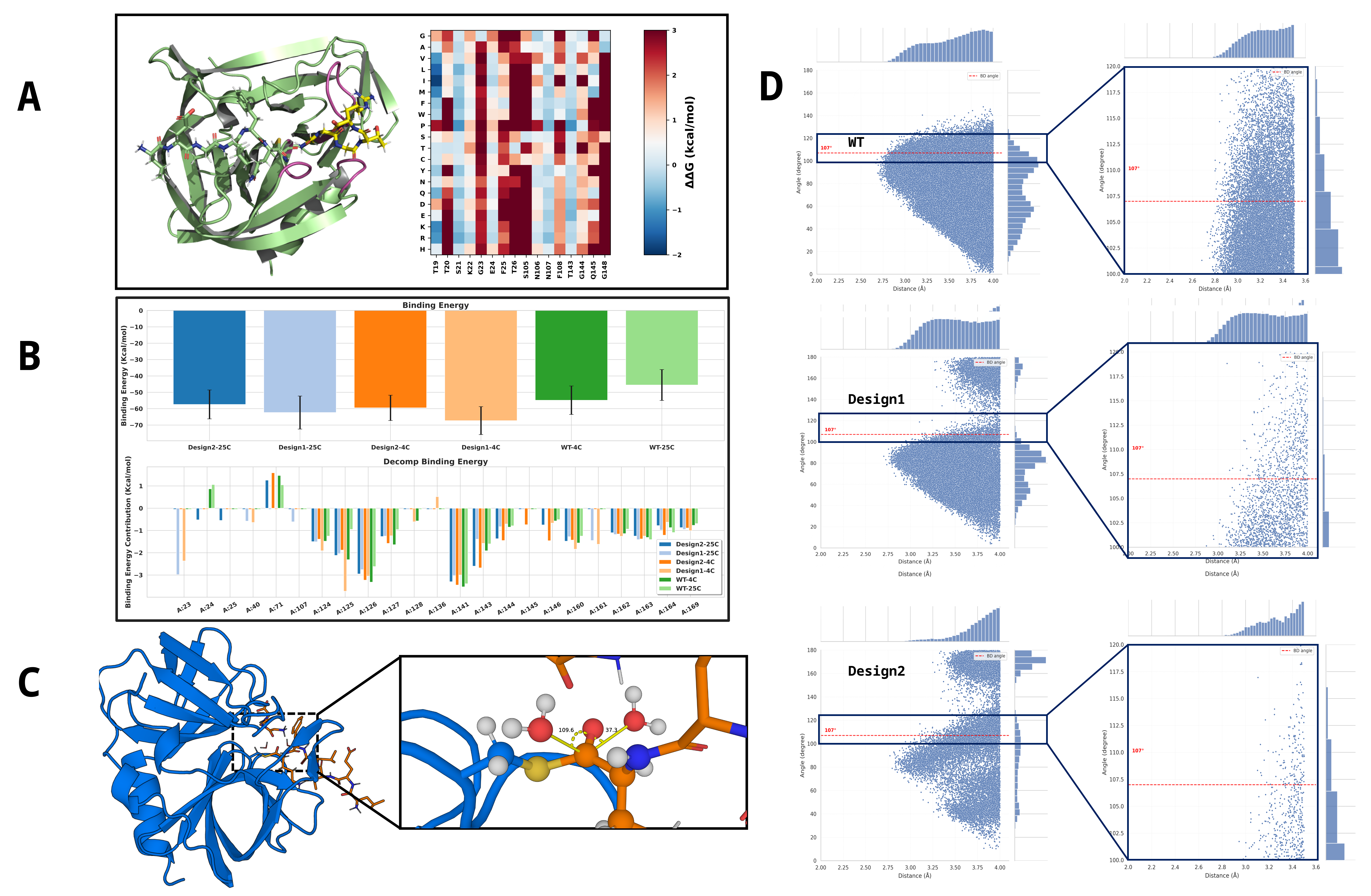
Figure 2. Utilization of PocketGen for pocket design and evaluation of binding and hydrophobic properties. (A) Results of the ΔΔG Scan for the Target Design Region. (B) Comparison of substrate binding energies for two high-order mutants and the wild-type(WT) enzyme at different temperatures. (C) Schematic diagram illustrating the attack angle formed between the thioester intermediate and surrounding water molecules. (D) Statistical distribution of the attack distance and attack angle for water molecules within 4 Å of the thioester intermediate. The red line indicates the canonical Bürgi-Dunitz (BD) angle. The statistical profiles of the two mutants exhibit significant differences compared to the WT.
To computationally identify mutations capable of converting HRV-3C protease into a peptide ligase, a three-tiered hierarchical screening protocol was implemented. The workflow was designed to first generate a targeted virtual library, then filter for variants that retain substrate binding capacity, and finally select candidates predicted to exhibit impaired hydrolytic activity. This strategy was based on the hypothesis that increased hydrophobicity of the active site would sterically disfavor the precise positioning of the nucleophilic water molecule required for hydrolysis of the acyl-enzyme intermediate.
A virtual library of HRV-3C variants was generated using the deep learning-based protein design tool PocketGen[22]. The AlphaFold3-predicted structure of the wild-type HRV-3C in complex with its hydrolytic substrate LEVLFQAPG was used as the design template. The design process was restricted to residues within 4-5 Å of the substrate’s C-terminal APG-binding groove, particularly those constituting the S1′ and S2′ pockets. A key constraint was introduced into the generative model to prioritize substitutions with hydrophobic amino acids (Val, Leu, Ile, Phe, Trp, Ala, Met), enforcing a statistical bias. In addition to using ESM[23] (evolutionary scale modeling)-2 15B throughout our experiments. Folding stability was ensured by screening mutation schemes based on a full-sequence saturation mutagenesis ΔΔG library, applying a threshold (Σ(ΔΔG) ≤ 2.0).
To ensure that the designed mutations did not disrupt essential substrate recognition, all variants from the generated library were subjected to a binding affinity screen. For each mutant, a non-covalent Michaelis complex was constructed in silico with its cognate peptide substrate LEVLFQGGG. Each complex was solvated in an OPC3 water box, neutralized with counterions, and subjected to three independent replicates of 100 ns cMD simulations using the AMBER ff19SB force field for binding conformation sampling. Subsequently, the binding free energy was calculated for each variant using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA)[24] method, analyzing snapshots from the final 80 ns of each MD trajectory. Only mutants exhibiting binding free energies comparable to or more favorable than that of the wild-type enzyme were advanced to the next stage.
The final screening stage aimed to identify candidates in which the geometry of the deacylation step is unfavorable for hydrolysis. For each variant that passed the binding affinity filter, a covalent thioester acyl-enzyme intermediate was modeled by forming a bond between the catalytic Cys146 and the carbonyl carbon of the P1 Gln residue of the substrate. The water attack properties are described by two parameters: The attack distance (Distance), defined by the distance between the nucleophilic water oxygen and the thioester carbonyl carbon; The water attack angle (Angle) ,defined by the angle formed by the nucleophilic water oxygen, the thioester carbonyl carbon, and the carbonyl oxygen. Those were sampled during three independent replicates of 100 ns classical MD simulations. Variants were prioritized if the statistical distribution of their BD angle showed a significant deviation from the canonical range considered optimal for nucleophilic attack (107° ± 7°). Candidates exhibiting a persistently non-optimal BD angle were selected as promising ligase designs for subsequent experimental validation.
The ligation reaction catalyzed by Trypsiligase were usually complete within minutes and requires 0.1 molar equivalents of enzyme with an excess of the corresponding acyl acceptor substrate (ligation substrate) because it needs to compete with the RH leaving group. Inspired by this, we propose to expand the pocket around the C-terminal peptide fragment released in the first process to facilitate its retention. The AlphaFold3-predicted structure of the wild-type HRV-3C in complex with its hydrolytic substrate LEVLFQAPG served as the design template. Around the C-terminus of the hydrolytic substrate, the original amino acid residues at positions 105-108 (gray region of the protein) underwent Motif Scaffolding via RFdiffusion[25], with the substrate C-terminal motif (APG) designated as Hotspots. This motif was extended to lengths of 8, 9, and 10 amino acids, generating 30 backbone structures for each design. Subsequently, using the hydrolytic substrate C-terminus (APG) as all-atom ligand information, a modified version of LigandMPNN[26] was employed to assign 200 sequences to each backbone structure, with a statistical bias applied towards hydrophobic amino acids. The design from each set with the highest overall_confidence was selected. The enzyme-substrate intermediate for this selected design was then subjected to 50 ns of cMD simulation at 4 °C and 25 °C using the AMBER ff19SB force field and the OPC3 water model for interaction sampling.
[11] E. H. Abdelkader and G. Otting, “NT*-HRV3CP: An optimized construct of human rhinovirus 14 3C protease for high-yield expression and fast affinity-tag cleavage,” Journal of Biotechnology, vol. 325, pp. 145–151, 2021, doi: https://doi.org/10.1016/j.jbiotec.2020.11.005.

This
study aimed to convert the HRV-3C protease, a highly specific cysteine hydrolase, into a novel
peptide ligase through rational design. Our primary strategy involved computationally engineering
a hydrophobic active-site pocket to suppress the enzyme’s intrinsic hydrolytic activity by sterically
hindering the access of nucleophilic water molecules.