In the vast landscape of biochemistry, transferases (EC 2) represent a major class of enzymes that catalyze the transfer of a functional group from one molecule (the donor) to another (the acceptor). Their catalytic activities are fundamental to metabolism and macromolecular synthesis in living organisms. For instance, glycosyltransferases are responsible for transferring activated sugar moieties to protein or lipid molecules, constructing intricate glycan networks that are crucial for cellular recognition and signaling. Similarly, kinases, which transfer phosphate groups, regulate the vast majority of cellular processes. Among all these transfer reactions, one of the most central and fundamental is the formation of the peptide bond. Within the magnificent molecular machine of the ribosome, the peptidyl transferase center catalyzes the sequential linkage of amino acids with remarkable precision and efficiency, synthesizing the proteins that form the basis of life’s functions.
The ability to precisely form peptide bonds is not only fundamental to life’s synthesis of proteins but also provides a powerful tool for biotechnology and chemical biology. Through controllable peptide transfer reactions (i.e., peptide ligation), researchers can achieve site-specific modification, labeling, cyclization, and fragment assembly of proteins, significantly expanding the scope of protein engineering and pharmaceutical development[1]. The fundamental prerequisite enabling these applications is specificity. An ideal peptide ligation tool must accurately recognize specific amino acid sequences to ensure the ligation reaction occurs exclusively at the predetermined site, avoiding non-specific modifications to other parts of the protein. This stringent requirement for specificity is an intrinsic property of many proteases. In the laboratory, tool enzymes such as Tobacco Etch Virus protease (TEV protease[2]), Human Rhinovirus 3C protease (HRV-3C protease[3]), and Thrombin are widely used for cleaving purification tags from fusion proteins. Their widespread adoption is precisely due to their high fidelity for specific recognition sequences (e.g., ENLYFQ/G for TEV, LEVLFQ/(G/P) for HRV-3C). This inherent high specificity provides a solid foundation for engineering them into ligases.
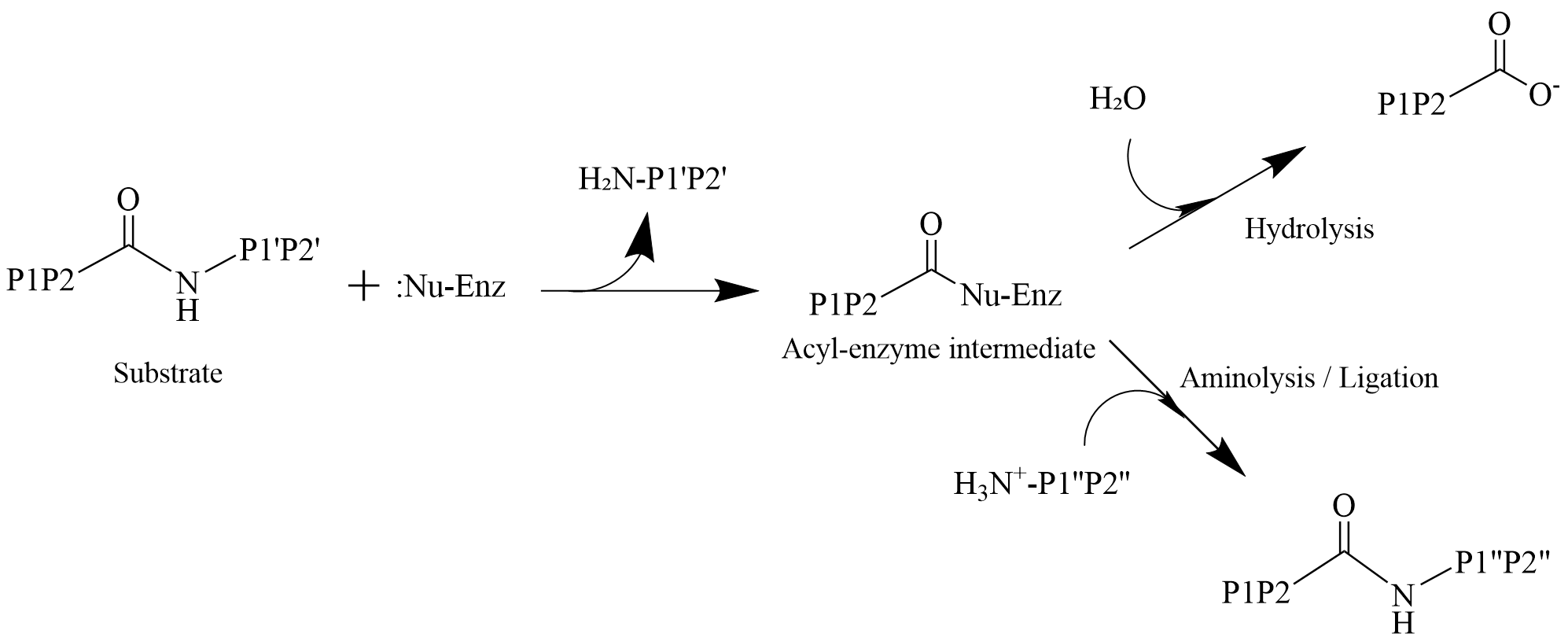
Figure 1. The two competing reaction pathways during the proteolytic stage.
The catalytic mechanisms of cysteine and serine proteases typically follow a two-step process. First, the nucleophilic residue of the enzyme (the thiol group of cysteine or the hydroxyl group of serine) attacks the carbonyl carbon of the substrate’s peptide bond. This results in the formation of a covalent acyl-enzyme intermediate and the release of the C-terminal peptide fragment. Subsequently, this intermediate is attacked by a second nucleophile, completing the catalytic cycle. This second step represents a critical “branching point.” In the conventional hydrolytic reaction, this nucleophile is a ubiquitous water molecule; its attack leads to the hydrolysis of the acyl-enzyme intermediate, releasing the N-terminal peptide fragment and regenerating the free enzyme. However, if an external nucleophile with a free N-terminal α-amino group (such as another peptide chain) is present, it can compete with the water molecule to attack the acyl-enzyme intermediate. Successful competition by this nucleophile results in the formation of a new peptide bond, accomplishing a peptide transfer (or ligation) reaction instead of hydrolysis.
The intrinsic specificity of proteases forms a foundation for engineering them into peptide ligases. Therefore, the “hydrolytic” and “ligation” activities of proteases are essentially two different outcomes resulting from the competition between different nucleophiles within the same catalytic pathway like Figure 1. However, under most physiological conditions, this reverse reaction involving nucleophilic attack by an amine on the ester intermediate is thermodynamically unfavorable. This process is governed by kinetic control. Strategies to shift the equilibrium of the protease-catalyzed reaction from hydrolysis towards ligation have been explored from multiple perspectives. These include altering the pH to modify the ionization states of reactive functional groups, employing organic co-solvents to reduce water activity, utilizing activated esters as substrates to accelerate nucleophilic attack, engineering proteases through site-specific modification or directed evolution, and combinations of the aforementioned approaches.
The idea of engineering proteases into peptide ligases has achieved remarkable success in practice, demonstrating the broad prospects of this approach. From serine proteases to ligases: the most classic example is the engineering of Subtilisin[4]. By mutating its catalytic serine to cysteine (S221C), supplemented by other key site mutations (such as P225A), scientists successfully developed the first-generation peptide ligase, Subtiligase[4]. This engineering strategy significantly reduced the hydrolysis rate while retaining the catalytic capability for aminonucleophiles, thereby enabling efficient peptide fragment ligation in aqueous solution. If the binding and reaction efficiency of the external nucleophilic peptide are simultaneously enhanced, it becomes possible to shift the reaction equilibrium toward the ligation product, thus transforming a natural “scissor” into a “glue”. A similar strategy was applied to the engineering of Trypsin, resulting in Trypsiligase[5], which exhibits stronger peptide aminolysis activity than hydrolysis and enables site-specific peptide linkage. In the design of enzymes such as Aqualigase[6], scientists promoted peptide condensation by weakening the efficiency of water molecules as nucleophiles, forming a hydrophobic reaction cavity that favors the release of water.
Nature provides perfect blueprints for this endeavor. For instance, Sortase A[7] from Staphylococcus aureus is a natural transpeptidase that specifically recognizes a C-terminal LPXTG motif and catalyzes its linkage to an N-terminal glycine residue. Furthermore, Butelase-1[8], a recently discovered peptide asparaginyl ligase from Clitoria ternatea, represents the most efficient peptide ligase known to date, whose remarkably high catalytic rate constant sets a new benchmark for enzymatic ligation. Asparaginyl Endopeptidases (AEPs), which also belong to the cysteine protease family, have been successfully engineered into powerful peptide ligation tools; these ligases are particularly useful for peptide head-to-tail cyclization and site-specific C-terminal protein labeling. Through targeted mutagenesis, their stringent substrate specificity has been relaxed while their ligation efficiency was optimized, leading to the development of a series of highly efficient enzymes known as Peptide Asparaginyl Ligases[9]. The successful engineering of AEPs[10] strongly validates the feasibility of developing ligase activity from cysteine proteases.
Based on the aforementioned background, this study aims to investigate strategies for converting a hydrolase into a ligase. We selected the HRV-3C as the engineering scaffold. As a cysteine protease, HRV-3C possesses several unique advantages: 1) Exceptionally high specificity: Its stringent sequence recognition provides a reliable foundation for precise ligation; 2) Well-established application protocols: It is already one of the most commonly used tool enzymes in biotechnology, with mature protocols for its expression, purification, and reaction conditions; 3) A unique recognition sequence: Its octapeptide recognition sequence is longer than those of many existing ligases, like the pentapeptide for Sortase A. Potentially offering higher addressing accuracy within complex protein substrates. Therefore, we hypothesize that via a rational design strategy, if the hydrolytic activity of HRV-3C can be suppressed while its peptide ligation activity is enhanced, a highly efficient peptide ligase with novel specificity could be created. The development of such a novel tool enzyme would provide a unique and powerful new option for site-specific protein conjugation, macromolecular drug synthesis, and chemical biology research.

[1] T. M. S. Tang and L. Y. P. Luk, “Asparaginyl endopeptidases: enzymology, applications and
limitations,” Org. Biomol. Chem., vol. 19, no. 23, pp. 5048–5062, 2021, doi: 10.1039/D1OB00608H.
[2] R. B. Kapust et al., “Tobacco etch virus protease: mechanism of autolysis and rational design of
stable mutants with wild-type catalytic proficiency,” Protein Engineering, vol. 14, no. 12, pp.
993–1000, 2001, doi: 10.1093/protein/14.12.993.
[3] X. Fan et al., “Quantitative Analysis of the Substrate Specificity of Human Rhinovirus 3C
Protease and Exploration of Its Substrate Recognition Mechanisms,” ACS Chemical Biology, vol.
15, no. 1, pp. 63–73, 2020, doi: 10.1021/acschembio.9b00539.
[4] A. M. Weeks and J. A. Wells, “Subtiligase-Catalyzed Peptide Ligation,” Chemical Reviews, vol.
120, no. 6, pp. 3127–3160, 2020, doi: 10.1021/acs.chemrev.9b00372.
[5] S. Liebscher et al., “N-Terminal Protein Modification by Substrate-Activated Reverse
Proteolysis,” Angewandte Chemie International Edition, vol. 53, no. 11, pp. 3024–3028, 2014, doi:
https://doi.org/10.1002/anie.201307736.
[6] Y. Feng et al., “Aqualigase: A Star Enzyme for One-Step Peptide Bond Dehydration
Condensation in a Nature Aqueous Phase,” ACS Catalysis, vol. 15, no. 13, pp. 11594–11607,
2025, doi: 10.1021/acscatal.5c01532.
[7] N. Suree et al., “The Structure of the Staphylococcus aureus Sortase-Substrate Complex Reveals
How the Universally Conserved LPXTG Sorting Signal Is Recognized*,” Journal of Biological
Chemistry, vol. 284, no. 36, pp. 24465–24477, 2009, doi: https://doi.org/10.1074/jbc.M109.022624.
[8] G. K. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian, and J. P. Tam, “Butelase 1 is an Asx-specific
ligase enabling peptide macrocyclization and synthesis,” Nature chemical biology, vol. 10, no. 9,
pp. 732–738, 2014.
[9] X. Hemu et al., “Structural determinants for peptide-bond formation by asparaginyl ligases,”
Proceedings of the National Academy of Sciences, vol. 116, no. 24, pp. 11737–11746, 2019, doi:
10.1073/pnas.1818568116.
[10] X. Hemu et al., “Turning an Asparaginyl Endopeptidase into a Peptide Ligase,” ACS Catalysis,
vol. 10, no. 15, pp. 8825–8834, 2020, doi: 10.1021/acscatal.0c02078.

This
study aimed to convert the HRV-3C protease, a highly specific cysteine hydrolase, into a novel
peptide ligase through rational design. Our primary strategy involved computationally engineering
a hydrophobic active-site pocket to suppress the enzyme’s intrinsic hydrolytic activity by sterically
hindering the access of nucleophilic water molecules.