Continuous Evolution
EvolvR System
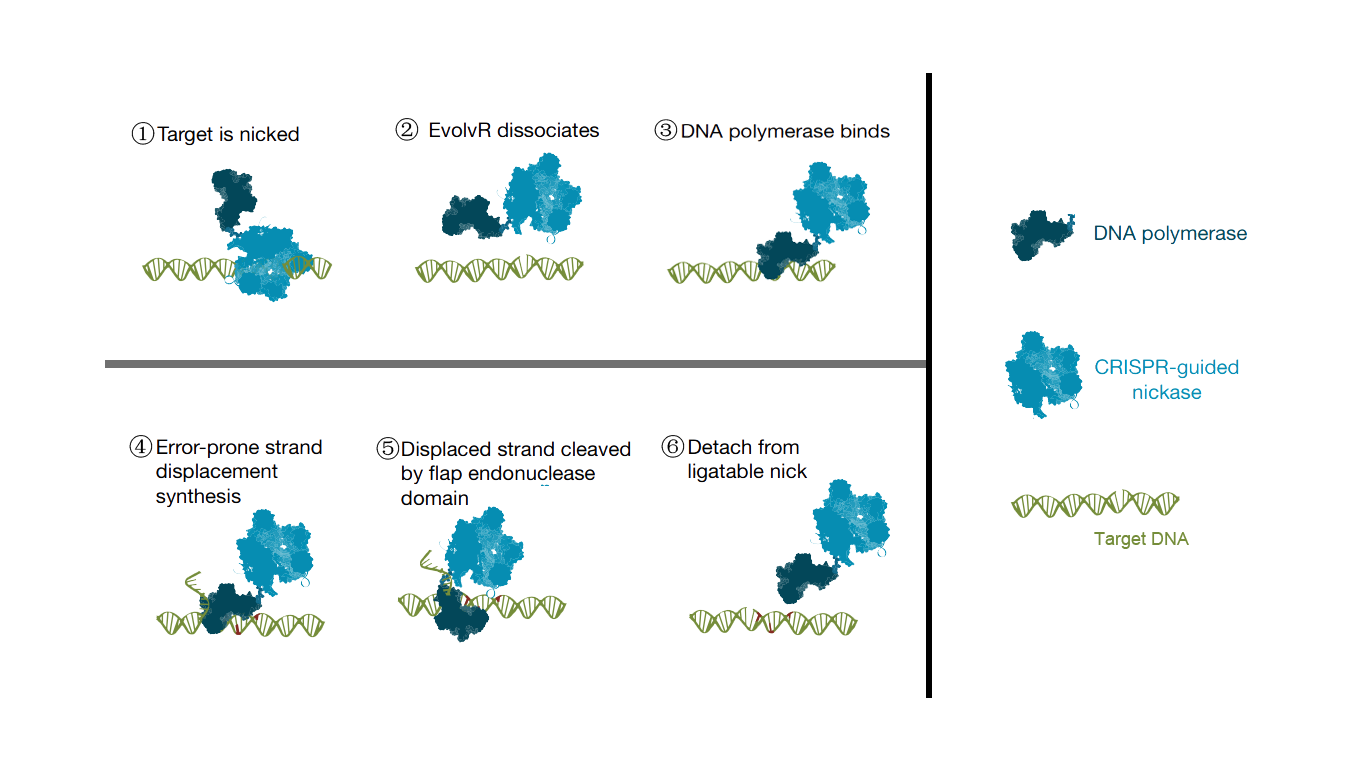
The EvolvR line combines the specificity of the CRISPR/Cas9 system with the mutagenic activity of error-prone DNA polymerases. This approach uses a designed guide RNA of about 20 nt to achieve precise targeting. EvolvR technology has two important components, including an enhanced nick Cas9 (enCas9) and an error-prone DNA polymerase I, which are fused together. These two proteins each have a number of point mutations to enhance their ability to cleave or make mutations. The mechanism of the EvolvR system is very simple, by gRNA, enCas9 is localized to the target motif, creating a gap. Later the error-prone DNA polymerase I fills the gap by randomizing the bases. This introduces a mutation in the target gene region.
We plan to construct EvolvR system in yeast for continuous directed evolution. Due to the long target gene (1059 bp),mutation on the whole genome is not realistic. On one hand, the screening time would be long, and on the other hand, it requires multiple gRNAs, and the selection tag of yeast is not enough. So, we analyzed the study of single point mutations for this enzyme and got its hotspot for mutation location, using one gRNA containing as many mutation sites as possible. And since the pEvolvR plasmid itself is a bacteria vector that can only be amplified in E. coli, we therefore planned to recombine it to the yeast expression plasmid YEGAP.
We plan to transfer three plasmids into Saccharomyces cerevisiae Hansen W303-1A for sequential directed evolution: a vector plasmid with the target gene (r-P415), a plasmid with the pEvolvR system (r-YEGAP) and a plasmid with sgRNA (r-P426). These three plasmids have different selection tags and are adapted to the defective phenotype of the yeast strain.
In order to achieve full gene coverage we had to design at least 4 gRNAs, but considering that the selection tag was not sufficient, we referred to other articles on directed evolutionary modification of this enzyme and analyzed the location of mutation hotspots that have an impact on function. A gRNA covering the above mutation hotspots was used in this work.
Scheme. 1. Principle of EvolvR.
We plan to construct EvolvR system in yeast for continuous directed evolution. Due to the long target gene (1059 bp),mutation on the whole genome is not realistic. On one hand, the screening time would be long, and on the other hand, it requires multiple gRNAs, and the selection tag of yeast is not enough. So, we analyzed the study of single point mutations for this enzyme and got its hotspot for mutation location, using one gRNA containing as many mutation sites as possible. And since the pEvolvR plasmid itself is a bacteria vector that can only be amplified in E. coli, we therefore planned to recombine it to the yeast expression plasmid YEGAP.
We plan to transfer three plasmids into Saccharomyces cerevisiae Hansen W303-1A for sequential directed evolution: a vector plasmid with the target gene (r-P415), a plasmid with the pEvolvR system (r-YEGAP) and a plasmid with sgRNA (r-P426). These three plasmids have different selection tags and are adapted to the defective phenotype of the yeast strain.
In order to achieve full gene coverage we had to design at least 4 gRNAs, but considering that the selection tag was not sufficient, we referred to other articles on directed evolutionary modification of this enzyme and analyzed the location of mutation hotspots that have an impact on function. A gRNA covering the above mutation hotspots was used in this work.
Plasmid Construction
Plasmids with TbsADH and its variants were synthesized by Sangon Biotech (Shanghai, China) and pEvolvR plasmid was from MiaoLingBio (Wuhan, China). P415,P426 and YEGAP were from Jionghong’s Lab. All Primers were synthesized by General Bio (Anhui, China). Plasmid extraction is performed using SanPrep Column Plasmid Mini-Preps Kit(Sangon Biotech, Shanghai, China). r-P415 was recombined from p415 (vector) and WT plasmid (insert), using primers p415-v-F/p415-v-R/WT-i-F/WT-i-R. r-YEGAP is recombinant from YEGAP (vector) and pEVOLVR plasmid (insert), using primers YEGAP-v-F/YEGAP-v-R/pEVolv-i-F/ pEVolv -i-R. r-P426 was created by targeted mutation of the p426 plasmid by homologous recombination using the primer p426-F/p426-R. PCR were conducted with Phanta® Max Super-Fidelity DNA Polymerase (Vazyme, Nanjing, China). DNA solutions were identified and isolated by 1% agarose gel electrophoresis (AGE). Homologous recombination was performed using DpnI (SibEnzyme, Beijing, China) and ClonExpress® II One Step Cloning Kit (Vazyme, Nanjing, China). After recombination, the recombinant products were first transferred into Escherichia coli DH5a competent cell, and the plasmids were re-extracted after sequencing was completed and then transformed into yeast strain with EX-Yeast Transformation Kit(Beijing Zoman Biotechnology Co.,Ltd.) .
Screening Strategy
It is well known that the bottleneck of directed evolution is the screening method. Without a good screening method, continuous directed evolution is no different from irrational design.
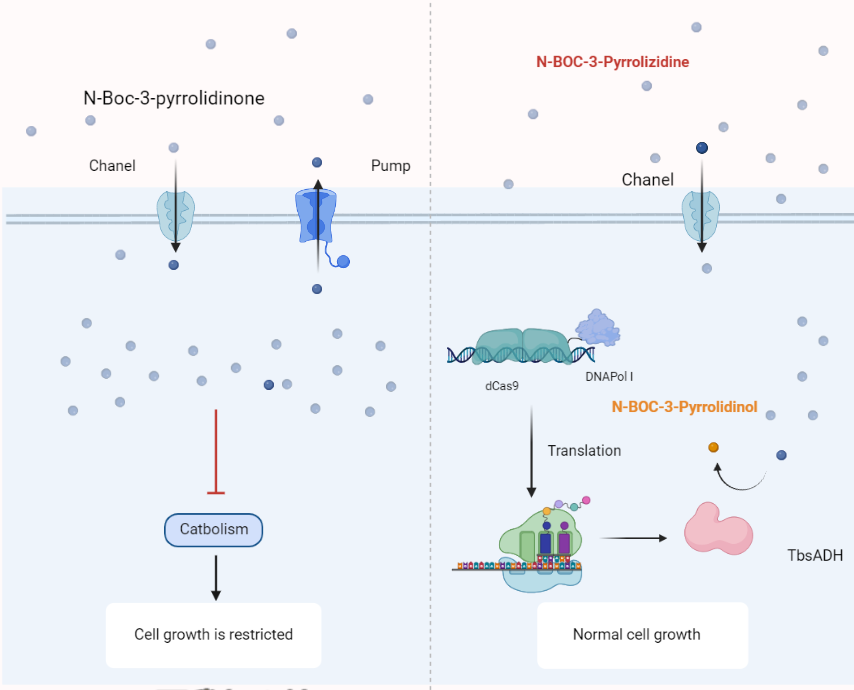
At the beginning of the subject design, we noticed the difference in the functional groups of the reactants and products. The first thing that comes to our mind when we see a heterocyclic compound is the problem of its toxicity. Coupling the enzyme activity to the cellular activity happens to be one of the most frequently used screening tools for directed evolution. The product NA carries a hydroxyl group that can be phosphorylated, which means that it can be made inaccessible to the cell by means of phosphorylation. In this way we have designed two universal directed evolution screening systems.
If both substrate and product are toxic, we can use surface display technology. The target protein is expressed on the surface of the yeast cell and a hydroxyl phosphorylase is coupled next to it. In this way the product will not be able to enter the cell due to electrostatic repulsion and will not have toxic effects on the cell.
If the substrate is toxic but the product is not, the display technique is not required.
Either way, yeast can be cultured using a solid or semi-solid medium containing the substrate. Spot the yeast to the center of the plate; the higher the target enzyme activity, the faster the yeast will grow. Pick the yeast that reaches the edge of the plate first for the next round of directed evolution. The reason why liquid medium is not used is because it can easily cause false positives.
Through pre-experiments we found that the substrate was toxic to yeast while the product was not, and there was no difference between the two for E. coli. So we used the latter means.
At the beginning of the subject design, we noticed the difference in the functional groups of the reactants and products. The first thing that comes to our mind when we see a heterocyclic compound is the problem of its toxicity. Coupling the enzyme activity to the cellular activity happens to be one of the most frequently used screening tools for directed evolution. The product NA carries a hydroxyl group that can be phosphorylated, which means that it can be made inaccessible to the cell by means of phosphorylation. In this way we have designed two universal directed evolution screening systems.
If both substrate and product are toxic, we can use surface display technology. The target protein is expressed on the surface of the yeast cell and a hydroxyl phosphorylase is coupled next to it. In this way the product will not be able to enter the cell due to electrostatic repulsion and will not have toxic effects on the cell.
If the substrate is toxic but the product is not, the display technique is not required.
Either way, yeast can be cultured using a solid or semi-solid medium containing the substrate. Spot the yeast to the center of the plate; the higher the target enzyme activity, the faster the yeast will grow. Pick the yeast that reaches the edge of the plate first for the next round of directed evolution. The reason why liquid medium is not used is because it can easily cause false positives.
Through pre-experiments we found that the substrate was toxic to yeast while the product was not, and there was no difference between the two for E. coli. So we used the latter means.
Scheme2: Schematic diagram of directed evolution using EvolvR.
Continuous Directed Evolution Process
The constructed engineered yeast was incubated in the defective liquid medium overnight. Configure semi-solid defective YPD medium containing NK concentrations of 1 g/L, 2 g/L, 3 g/L, and 5 g/L, respectively. Dilute the engineered yeast broth 300 times and then take 50 μl and apply it to the center of the plate with 1 g/L NK concentration. After overnight incubation for 3 days, pick the marginal yeast strain in liquid defective YPD medium and then inoculate to the center of the plate at 2 g/L. And so on. Samples were left for each pick for enzyme activity assay and plasmid extraction for sequencing.
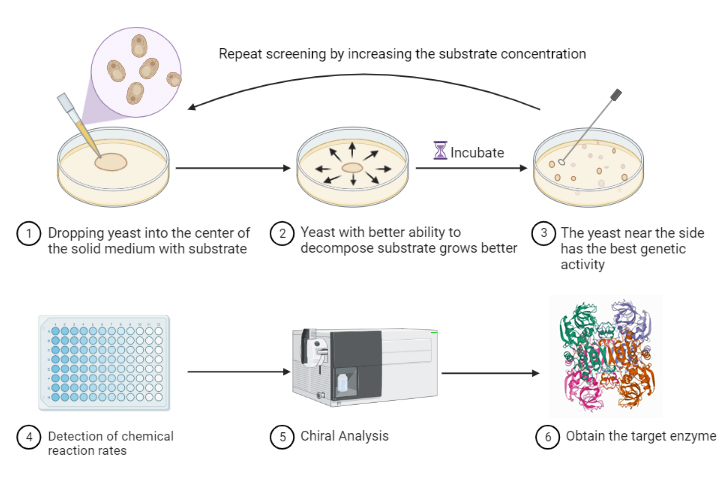
Scheme3: Continuous directed evolutionary process.
Enzyme Property Identification
Alcohol Dehydrogenase Assay Kit (Solarbio, Beijing, China) were used to initially determine enzyme activity after large-scale protein expression, including bacterial fragmentation supernatant and pure enzyme.
Recombination and expression of glucose dehydrogenase
In order for the reactions to constitute a cycle, we coupled the main reaction to a glucose dehydrogenation reaction for the recycling of NADPH. The reaction process can also be judged industrially by the consumption of sodium carbonate buffer by the gluconate generated. To achieve this reaction coupling, we extracted Bacillus subtilis WB800N genome and recombine the glucose dehydrogenase(GDH) onto pET21a plasmid. We then used E. coli BL21 for expression and took the cell crushing supernatant for reflective system construction.
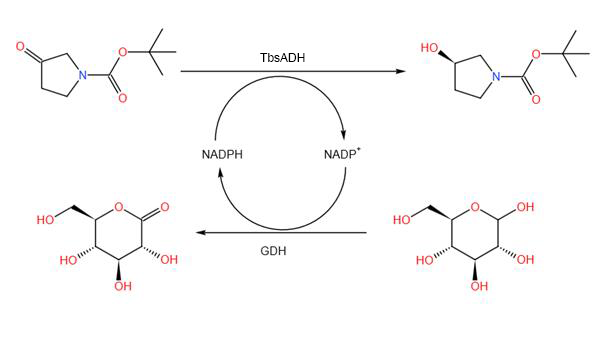
Scheme. 4. Chemical reaction diagram
Construction of the reaction NK-NA reaction system
Specific reaction systems include 18 ml PBS, 10 mM ZnCl2, 10 mM MgCl2, 2gNK, 2.5 mg NADPH, 2.5 g Glucose, 1 ml GDH and 1 ml TbsADH.
The yield was detected using Gas chromatography mass spectrometry(GCMS) and its chiral selectivity was detected by Ultra Performance Liquid Chromatography(UPLC).
The yield was detected using Gas chromatography mass spectrometry(GCMS) and its chiral selectivity was detected by Ultra Performance Liquid Chromatography(UPLC).
Next :Results Page