




The sensor comprises a catalytic module, NdmB, and a sensor module, the pYB1s-ndmDCEA plasmid. Bacteria can only survive when NdmB catalyzes caffeine to produce paraxanthine (PX), and pYB1s-ndmDCEA degrades PX into xanthine. To validate the effectiveness of the sensor system, the pYB1s-ndmDCEA plasmid was introduced into an Escherichia coli (E. coli) strain with the guaB gene knocked out. Control experiments revealed that the strain showed optimal growth in media containing xanthine and PX, in contrast to media supplemented with theobromine (Tb), theophylline (Tp), caffeine, or media without additional xanthine compounds. These results confirmed the necessity of PX for bacterial growth (Figure 1A). Moreover, different concentrations of PX were tested to observe bacterial growth, and a positive correlation between growth and PX concentration was observed, demonstrating that the sensor successfully detects and utilizes PX (Figure 1B).
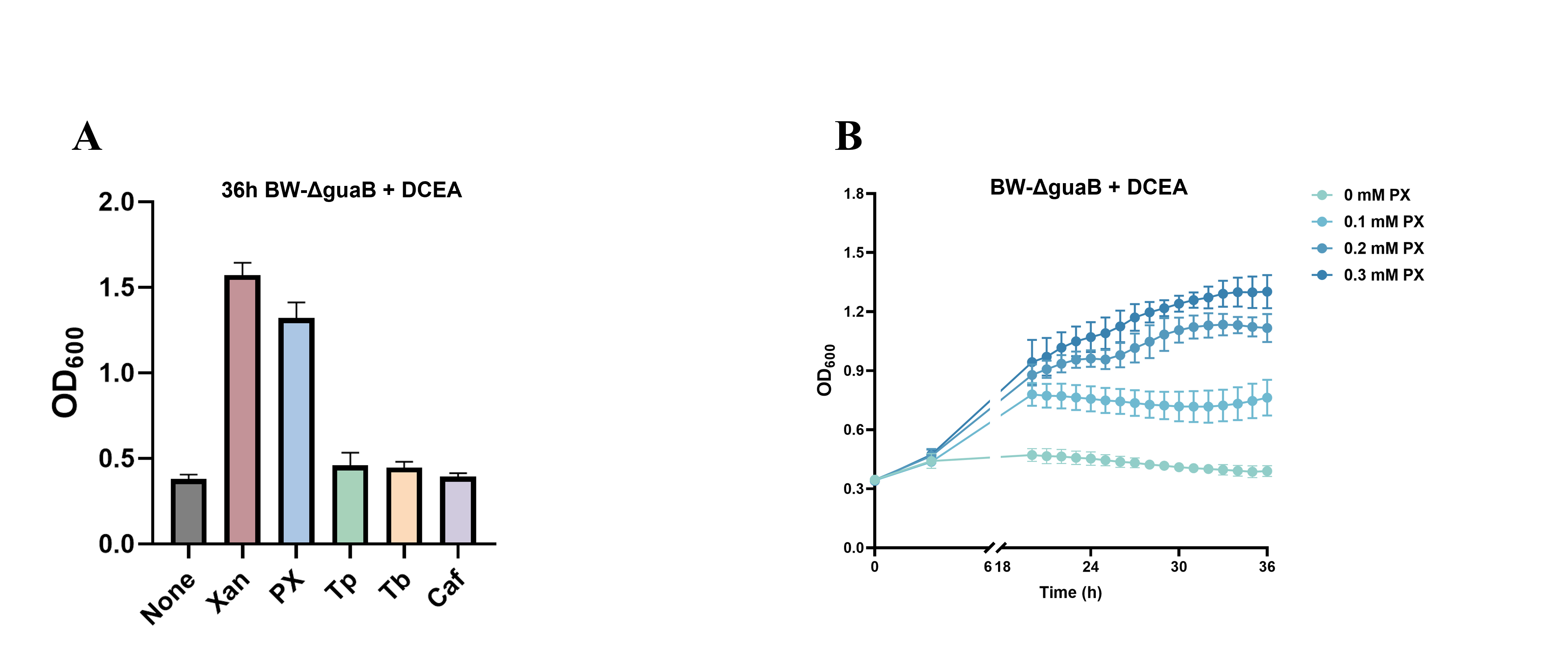
Figure 1: Validation of the whole-cell biosensor for PX. (A) OD600 values of bacterial growth under different substrate conditions. The most significant growth was observed in media containing PX. (B) The effect of PX concentration on bacterial growth. A positive correlation between bacterial growth and PX concentration demonstrates that the sensor successfully detects and utilizes PX.
After confirming the functionality of the whole-cell biosensor, we used it to screen NdmB mutants with enhanced caffeine degradation capabilities. Based on previously reported key sites, we performed combinatorial mutations at two sets of sites (Q289 and L293, and W256, C267, and M271) and screened the viable strains using the sensor plasmid (Figure 2).
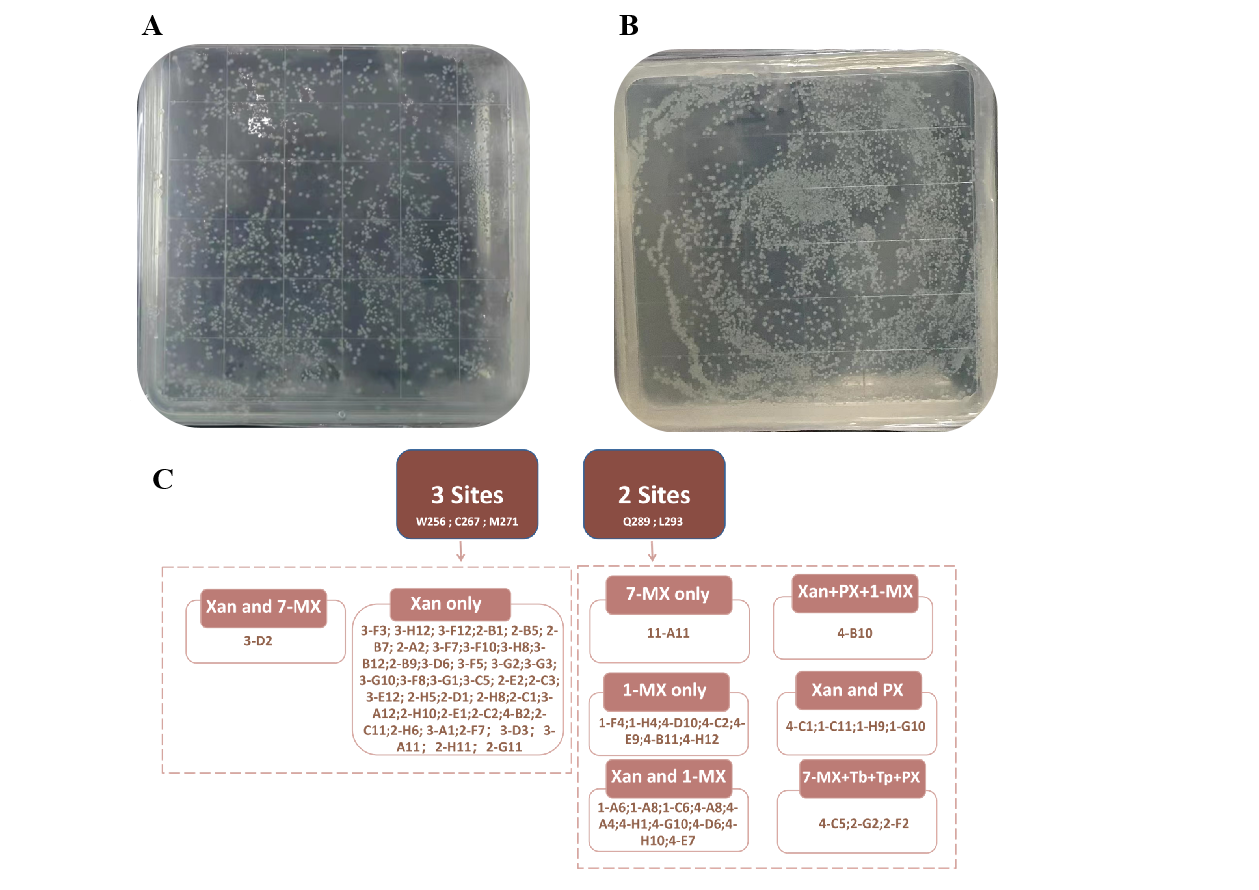
Figure 2. NdmB combinatorial mutations. (A) Mutations at positions Q289 and L293. (B) Mutations at positions W256, C267, and M271. (C) This figure summarizes the mutation types and their corresponding products. Strains with higher OD600 values produce not only hypoxanthine but also other methylxanthine compounds (e.g., 1-MX and 7-MX). Among the mutants, those that directly produce xanthine constitute the majority.
During the experiment, E. coli strains carrying the mutant ndmB gene were inoculated into the medium, and after inducing protein expression, the supernatant was removed by centrifugation. Then, a caffeine-containing transformation solution prepared with 1X M9 buffer was added for whole-cell catalysis. After the catalysis was completed, 200 µL of the supernatant was transferred to a 96-well plate, and the whole-cell biosensor was added. By monitoring the growth of E. coli with the guaB gene knocked out, we screened strains with higher OD600 values (Figure 3). The reaction products were then detected using high-performance liquid chromatography (HPLC) and the strains were sequenced for validation.
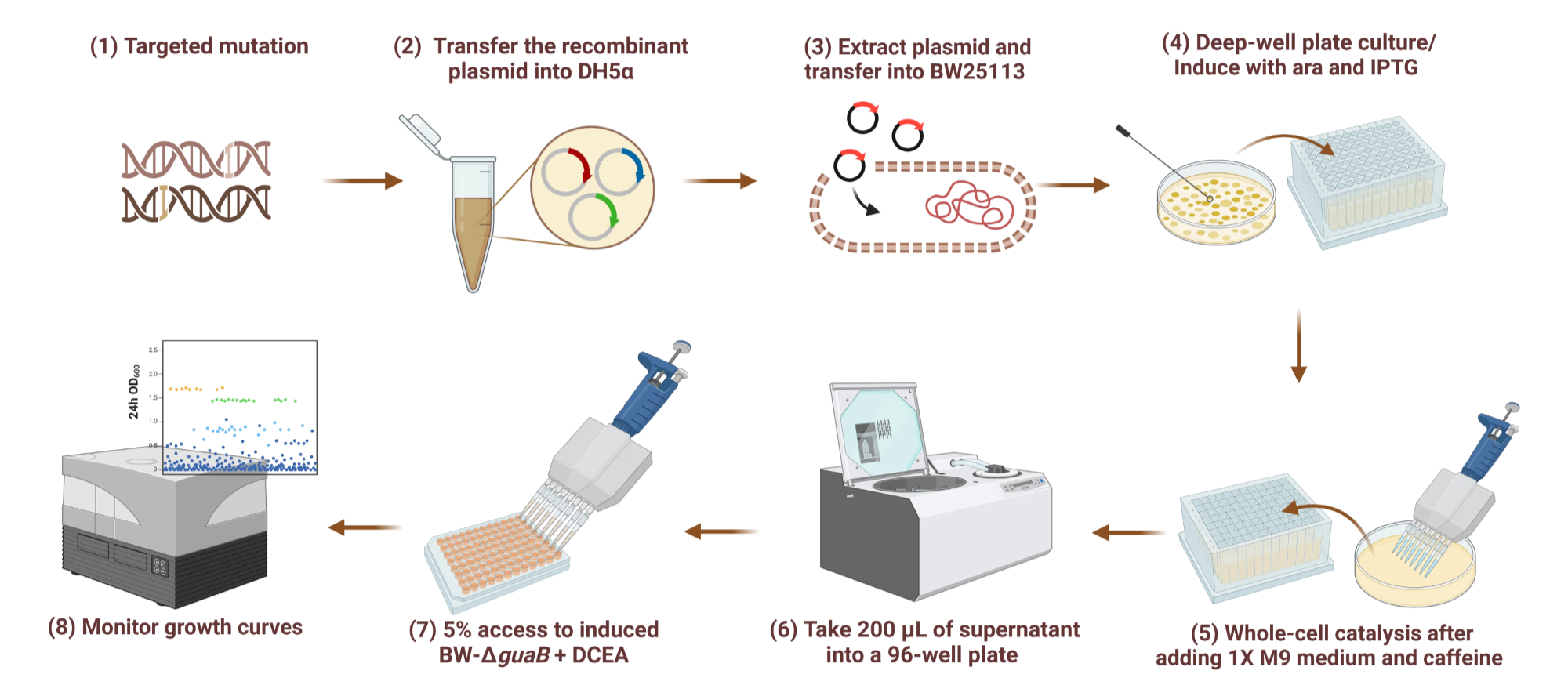
Figure 3. Schematic diagram of the directed evolution and mutant screening process for NdmB.The process includes: (1) Site-directed mutagenesis of key residues in NdmB using degenerate primers; (2) Transformation of recombinant plasmids into E. coli DH5α competent cells for amplification; (3) Plasmid extraction and transformation into the E. coli BW25113 strain; (4) Induction of mutant NdmB expression in deep-well plates containing arabinose and IPTG; (5) Addition of 1 × M9 medium and caffeine for whole-cell catalysis; (6) Collection of the supernatant by centrifugation, transfer to a 96-well plate; (7) Addition of the whole-cell biosensor; (8) Monitoring of bacterial growth curves to evaluate the catalytic efficiency of each mutant in producing PX.
After conducting HPLC to detect the reaction products and DNA sequencing to determine the mutant sequences, we analyzed the mutation types and their associated products . Surprisingly, the strains with high OD600 values produced not only PX but also other methylxanthine derivatives, such as 1-MX and 7-MX. These compounds were also converted into xanthine in the sensing module, allowing the strains to survive. This indicates that NdmB mutants gained demethylation activity not only at the N3 position but also at the N1 and even N7 positions (Figure 4). The diversity of products generated by these NdmB mutants significantly increased the complexity of our screening, and further investigation was necessary to identify mutants that could selectively promote PX production.
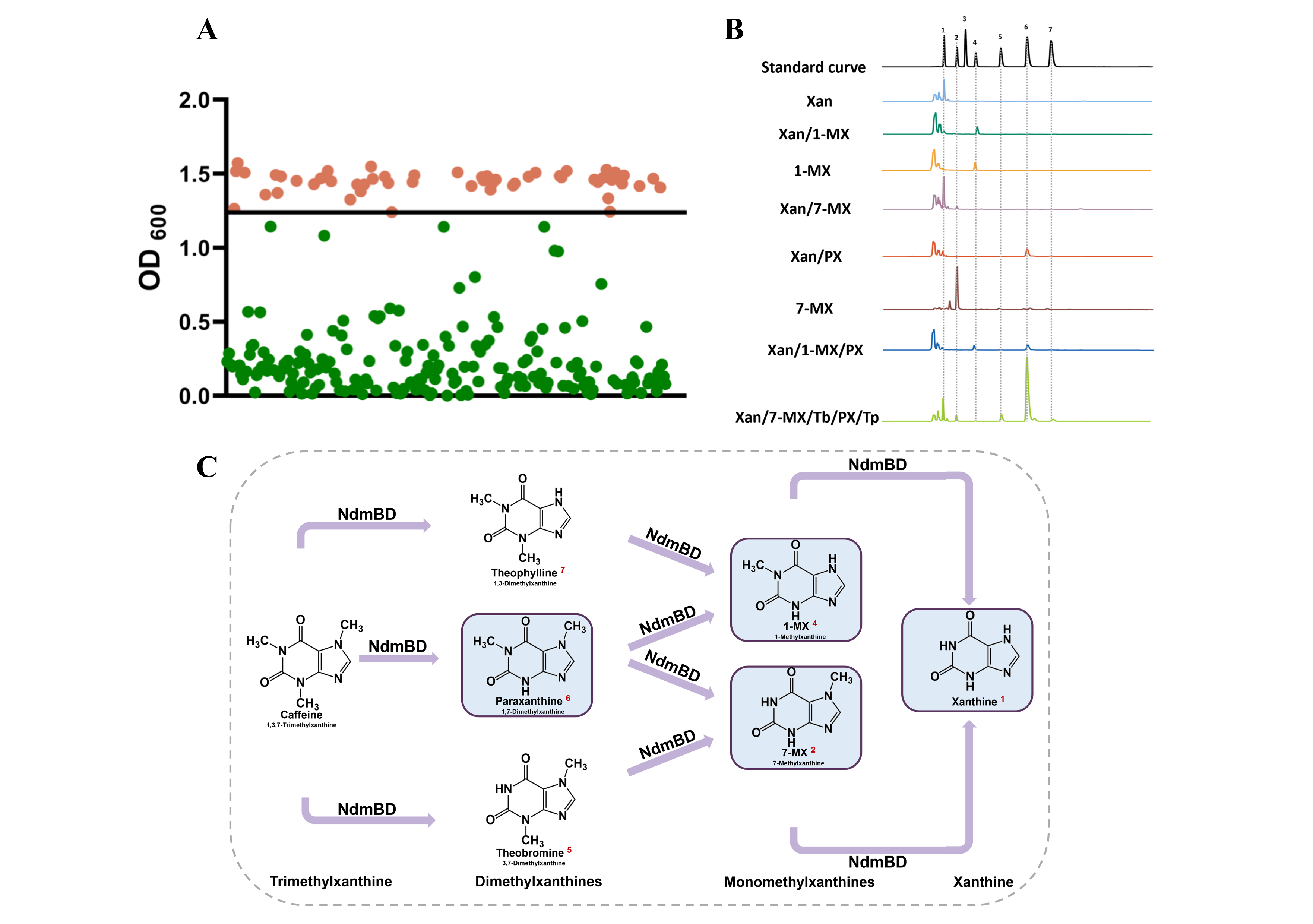
Figure 4. Screening results and by-product analysis of NdmB mutants.(A) Screening results of mutants using the PX whole-cell biosensor. The figure highlights positive clones with higher OD600 values (in orange), indicating that these strains likely have a stronger capability for PX production. (B and C) HPLC analysis of the standard curves for products from different strains. The analysis confirmed that the mutants produced not only PX but also other demethylated by-products, such as 1-MX and 7-MX.
The integration of machine learning with evolutionary engineering can significantly enhance the efficiency and effectiveness of protein optimization1. To identify mutation sites favoring PX production, we employed a Ridge regression model for machine learning analysis. Analysis of the regression coefficients revealed that mutations at Q289 showed a strong positive effect on the synthesis of both PX and 7-MX, while their impact on other byproducts was relatively minor. The experimental results provide critical insights for subsequent site-directed mutagenesis experiments at Q289 (Figure 5).
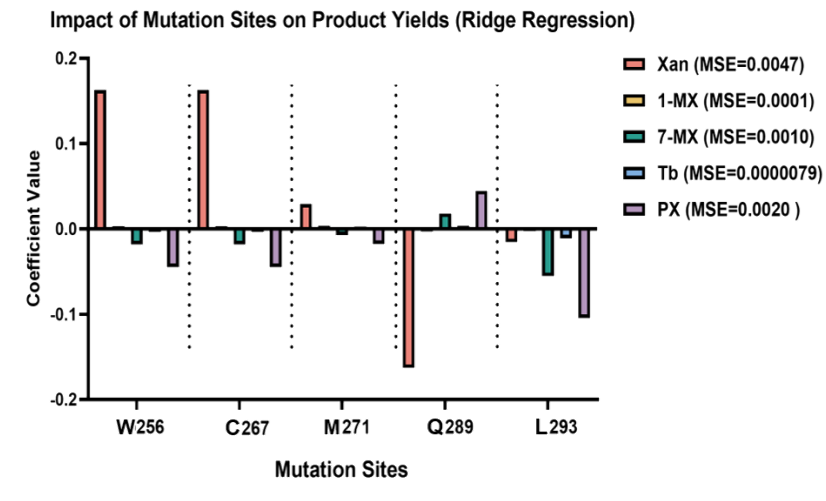
Figure 5. The Ridge regression model analyzed the influence of five mutation sites (W256, C267, M271, Q289, L293) on the generation of different products. Positive regression coefficients indicated that the mutations had a favorable effect on methylxanthine production, with Q289 mutations significantly enhancing the production of PX and 7-MX, while other sites tended to promote the formation of different demethylated products.
Based on the predictions from the machine learning model, we designed degenerate primers to perform site-directed saturation mutagenesis at Q289. The mutants were screened using the whole-cell biosensor, and strains with higher OD600 values were selected for further analysis through HPLC and DNA sequencing. The experimental data revealed two key findings: first, when Q289 was mutated to threonine(NdmBQ289T), PX production increased significantly, reaching 1.22 g/L (6.76 mM), which is 7.3 times higher than the previously reported maximum yield. Second, as the side chain (R-group) decreased in size (threonine → serine → alanine), a distinct shift in the types and ratios of products was observed: smaller R-groups resulted in a gradual decrease in PX production, while the yield of 7-MX increased accordingly. This pattern indicates that the size of the R-group at the position 289 significantly influences the enzyme's substrate selectivity: as the R-group becomes smaller, NdmB not only catalyzes N3-demethylation but also exhibits N1-demethylation activity (Figure 6).
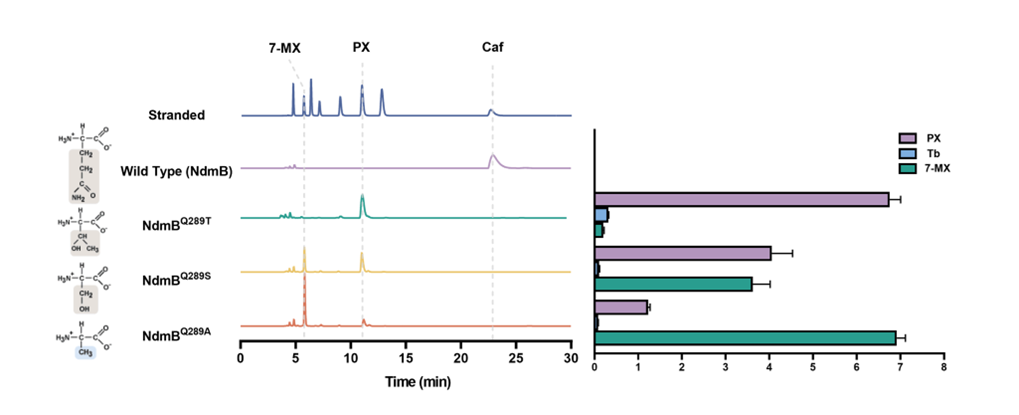
Figure 6. HPLC analysis of catalytic products from NdmB mutants. The NdmBQ289T mutant exhibited a significant increase in PX production, while the NdmBQ289A mutant showed a preference for producing 7-MX. Larger R-groups (such as threonine) favor PX production, while smaller R-groups (such as alanine) tend to generate 7-MX.
Site-directed mutagenesis may have a profound impact on the selection of enzymes with desired activities2. To precisely simulate the behavior of the target protein under whole-cell catalysis conditions, we optimized the NdmB structure obtained from the PDB using molecular dynamics (MD) simulations. MD simulations allow us to track the conformational dynamics of proteins at the atomic level. After generating the models for wild-type NdmB and its mutants (NdmBQ289T, NdmBQ289S, and NdmBQ289A), we performed molecular docking of caffeine with NdmB . The interactions between NdmB and methylxanthine structures, including steric hindrance and hydrogen bonding, jointly regulated substrate positioning within the active site.
The docking results indicated that the steric hindrance of Q289 affected PX production (Figure 7A and 7B). Furthermore, when PX was used as a substrate, its carbonyl group interacted with the Thr residue, preventing PX from entering the active pocket further. This simulation likely explains the NdmBQ289T mutant's specificity for N3-demethylation (Figure 7C). In the NdmBQ289S mutant, once PX entered the activesite, a part of it was further converted to 7-MX, while the rest remained unreacted due to steric hindrance from the Ser residue's flexible side chain (Figure 7D and 7E). In the NdmBQ289A mutant, PX did not interact with NdmBQ289A, allowing the N1-methyl group of PX to approach closer to the active center, thereby increasing 7-MX production (Figure 7F). Overall, the site-directed saturation mutagenesis of Q289 significantly produced multiple mutants with different efficiency of demethylation reactions, which offered valuable insights for future enzyme engineering.
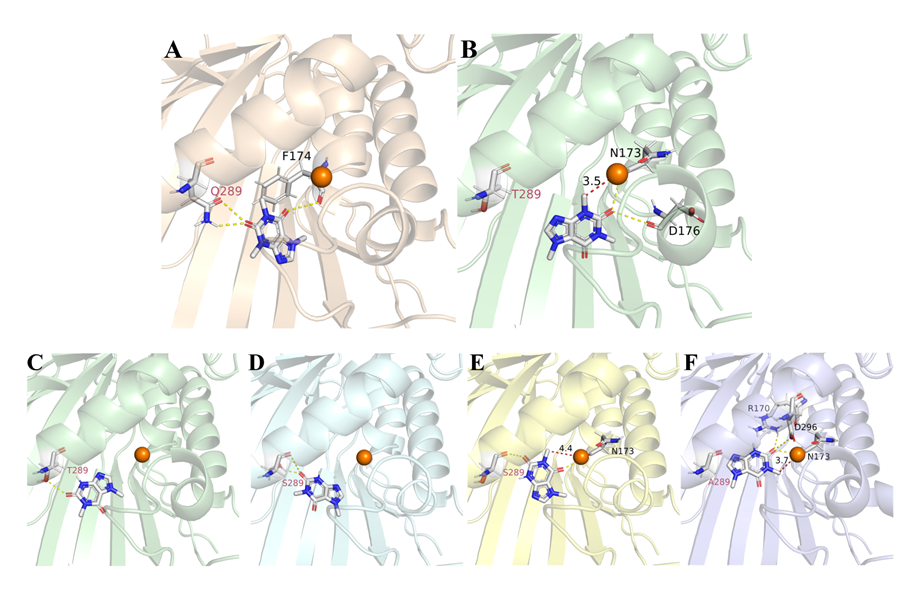
Figure 7. Molecular docking analysis of NdmB mutants with substrates. (A) Docking analysis of wild-type NdmB with caffeine, showing that the key residue NdmBQ289T blocks caffeine from entering the active pocket. (B) Docking analysis of NdmBQ289T with caffeine indicates that reduced steric hindrance allows the N3 methyl group of caffeine to approach the active center at a distance of 3.5 Å. (C) Docking analysis of NdmBQ289T with PX reveals that the absence of the N3 methyl group exposes a carbonyl group that interacts with the Thr residue, which prevents further demethylation. (D and E) Docking analysis of NdmBQ289S with PX demonstrates steric effects from the Ser side chain,partially converting PX to 7-MX. (F) Docking analysis of NdmBQ289A with PX indicates that lack of A289 interaction with PX allows the N1 methyl group to approach the active center, leading to increased production of 7-MX.
We conducted molecular dynamics simulations of wild-type NdmB and the NdmBQ289T mutant. The results showed that over time, the distance between the N3-methyl group and the non-heme iron in both the wild-type and mutant proteins became similar in the later stages of the simulation, indicating that proximity to the non-heme iron is not the sole determinant of wild-type reactivity (Figure 8A). RDF (radial distribution function) analysis revealed that the RDF profiles of the wild-type and mutant were generally similar. However, the mutant exhibited a slightly higher peak and slower decline, suggesting that the mutation induced local structural changes, resulting in stronger interactions with surrounding molecules or a higher distribution density, leading to a more stable binding between the mutant and caffeine (Figure 8B). PCA (principal component analysis) of vec1 showed significant differences between the wild-type and mutant, reflecting different motion patterns over the simulation time and indicating distinct overall motion trajectories (Figure 8C). The regions of positive covariance in the wild-type were relatively sparse, indicating less coordination in the overall motion of NdmB. In contrast, the positive covariance values between atoms in the mutant were more concentrated, and the areas of negative covariance in NdmBQ289T were reduced, suggesting that the atoms in the mutant exhibited more coordinated and focused movements during the simulation, which could imply greater structural stability or more efficient binding with the caffeine substrate (Figures 8D and 8E). Analysis of the atoms near caffeine revealed that the mutation affected the openness of the active site. Compared to the wild-type, the mutant displayed a smaller interaction area (Figure 8F). This supports the “two-pocket” hypothesis, which proposes that the mutation creates a more confined pocket, better positioning caffeine for interaction with the non-heme iron, whereas the wild-type interacts with more residues. In the wild-type active site pocket, the methyl group of caffeine is blocked by surrounding residues, preventing direct contact with the non-heme iron (Figure 8G). In the mutant, caffeine is positioned more favorably, with reduced spatial hindrance, allowing better alignment of the methyl group with the non-heme iron (Figure 8H). This confirms the “two-pocket” hypothesis, indicating that the mutant creates a more direct pathway for interaction with the non-heme iron.
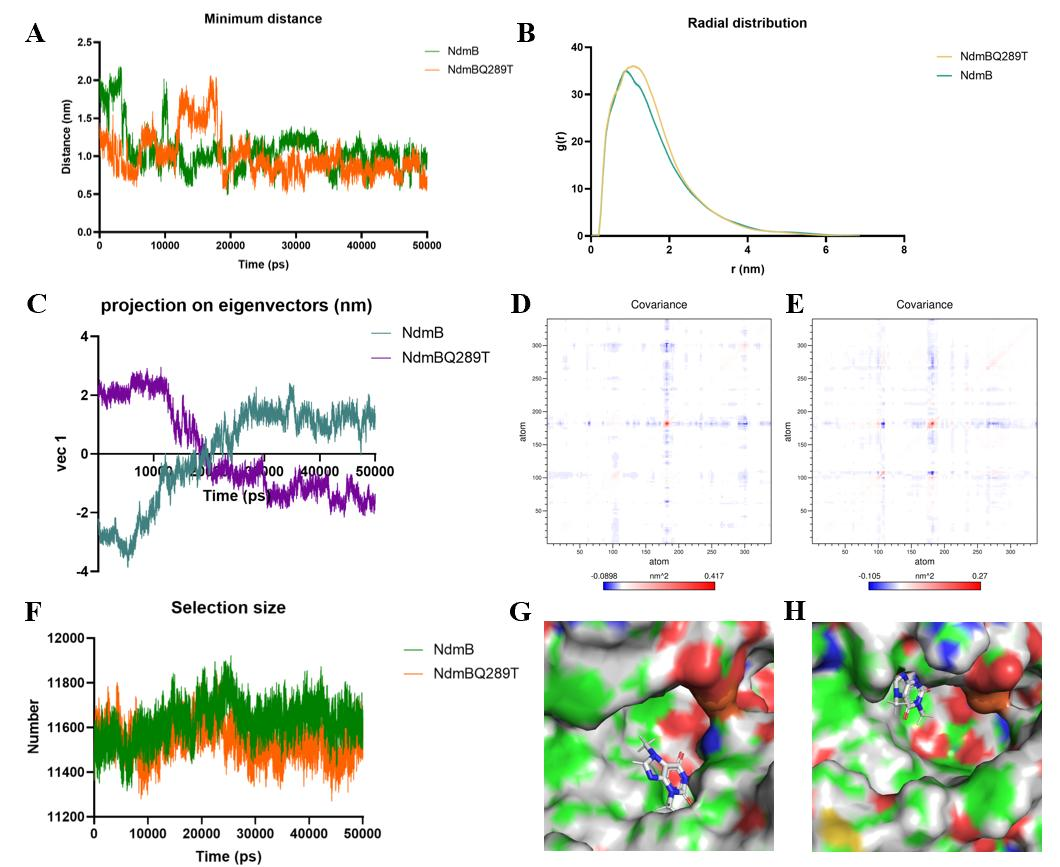
Figure 8. Analysis of caffeine binding in wild-type NdmB and mutant NdmBQ289T. (A) Analysis of the distance between the N3-methyl group and non-heme iron. (B) Radial distribution function (RDF) analysis: the RDF profiles of the wild-type and mutant are generally similar. (C) Principal component analysis (PCA) projection of feature vectors. (D) Covariance analysis of wild-type NdmB. (E) Covariance analysis of the mutant: in comparison, the mutant shows more concentrated positive covariance, and the areas of negative covariance in NdmBQ289T are reduced, indicating that atomic movement is more consistent and coordinated throughout the simulation, potentially contributing to increased structural stability. (F) Analysis of atoms near caffeine. (G) Visualization of the binding pocket in wild-type NdmB. (H) Visualization of the binding pocket in NdmBQ289T.
The polar repulsion and hydrogen bond interactions between NdmB and the structure of methylxanthine together regulate the positioning of the substrate at the enzyme's active site. We further conducted a hydrogen bond analysis of wild-type NdmB and the mutant NdmBQ289T. The results showed that hydrogen bonds in wild-type NdmB primarily formed between 5000–7500 ps and 8500–10000 ps (Figure 9A). During the first 10000 ps, the amino hydrogen on the R-group of NdmBQ289T formed hydrogen bonds multiple times with the carbonyl group of caffeine, and the carbonyl groups frequently generated repulsive forces, preventing caffeine from approaching the active site (Figure 9B). Since caffeine could not reach the active site, it shifted in another direction and eventually bound to a nearby pocket region (5568 ps), where it stabilized. However, this pocket was separated from the non-heme iron in the active center by other amino acid residues, making it difficult for caffeine to contact the non-heme iron at this location (Figure 9C). In contrast, hydrogen bonds in the mutant NdmBQ289T were easily stabilized after 40000 ps (Figure 9D). NdmBQ289T rarely formed hydrogen bonds with caffeine, and the spatial hindrance was minimal, allowing caffeine to enter the pocket smoothly and approach the active site near the non-heme iron. Once caffeine entered the active site, the amino hydrogen on residue R170 formed hydrogen bonds with caffeine's carbonyl group, preventing it from moving away from the non-heme iron (Figure 9E). Besides not obstructing caffeine from entering the pocket, NdmBQ289T could also generate repulsive forces when caffeine attempted to leave the pocket, thereby hindering its exit (Figure 9F).
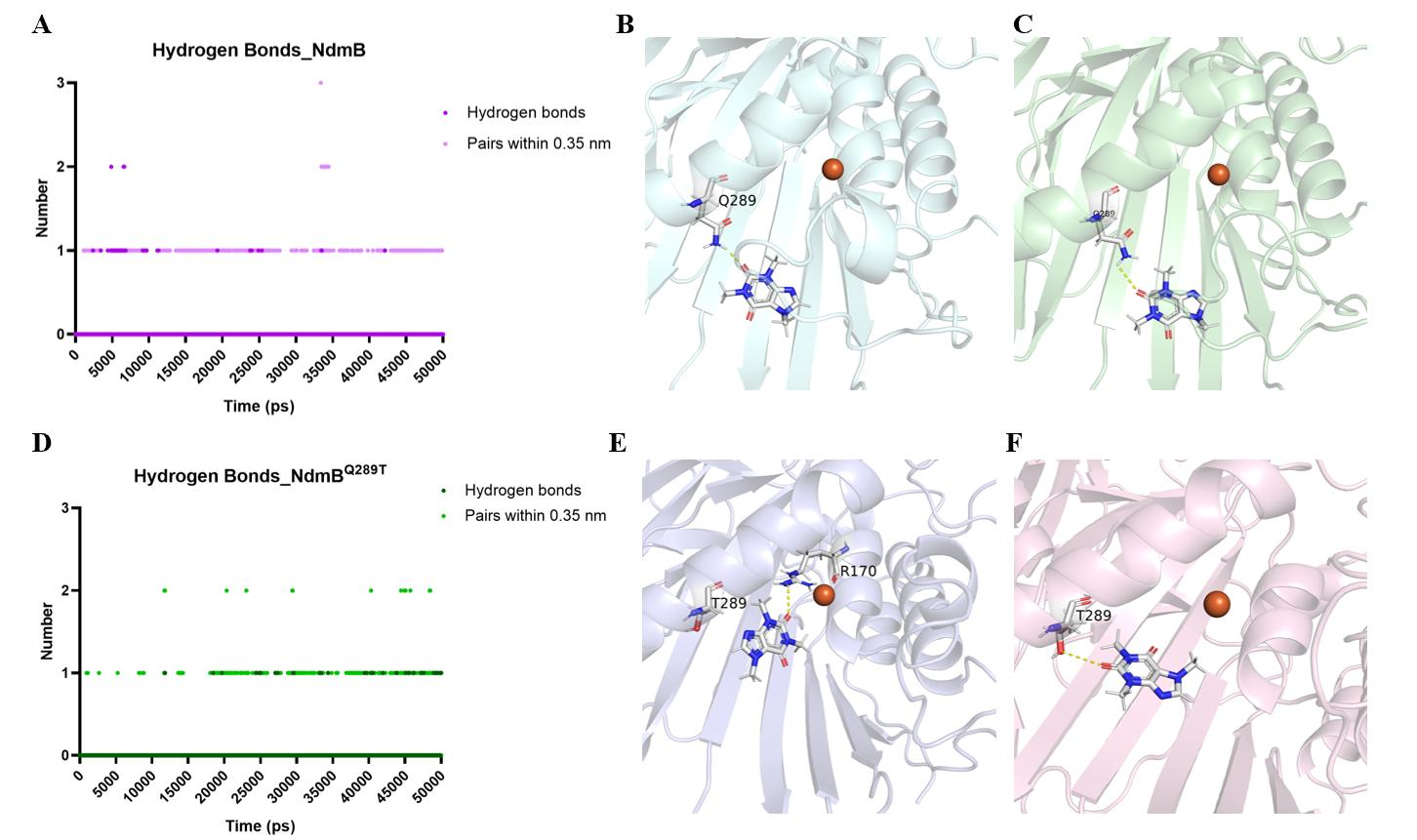
Figure 9. Hydrogen bond analysis. (A) Hydrogen bonds in wild-type NdmB. (B) Structure of wild-type NdmB with caffeine at 5202 ps: the amino hydrogen of Q289 forms hydrogen bonds multiple times with the carbonyl group of caffeine, preventing caffeine from approaching the active site. (C) Structure of wild-type NdmB with caffeine at 5568 ps: caffeine cannot approach the active site and shifts to bind in another pocket region, but is unable to contact the non-heme iron. (D) Hydrogen bonds in mutant NdmBQ289T. (E) Structure of mutant NdmBQ289T with caffeine at 45060 ps: 289T forms almost no hydrogen bonds with caffeine, allowing caffeine to smoothly enter the pocket and approach the non-heme iron. (F) Structure of mutant NdmBQ289T with caffeine at 48410 ps.
Promoter optimization is a key strategy to improve metabolic pathways, particularly the -10 region, which plays a crucial role in RNA polymerase binding and transcription initiation3. In this study, we performed random mutagenesis in the six bases of the -10 region of the pJ23107 promoter, followed by screening using a wholecell biosensor. Next, we selected the strains with the highest activity for larger-scale fermentation and analyzed their methylxanthine production. The results demonstrated that under conditions of 200 OD and 40 mM substrate concentration, the PX yield of the E8 strain reached 5.42 g/L, which represents a 4.4-fold increase compared to the NdmBQ289T mutant without metabolic engineering, and a 29-fold improvement over the highest reported PX yield to date. These findings highlight the success of optimizing metabolic pathways (Figure 10).
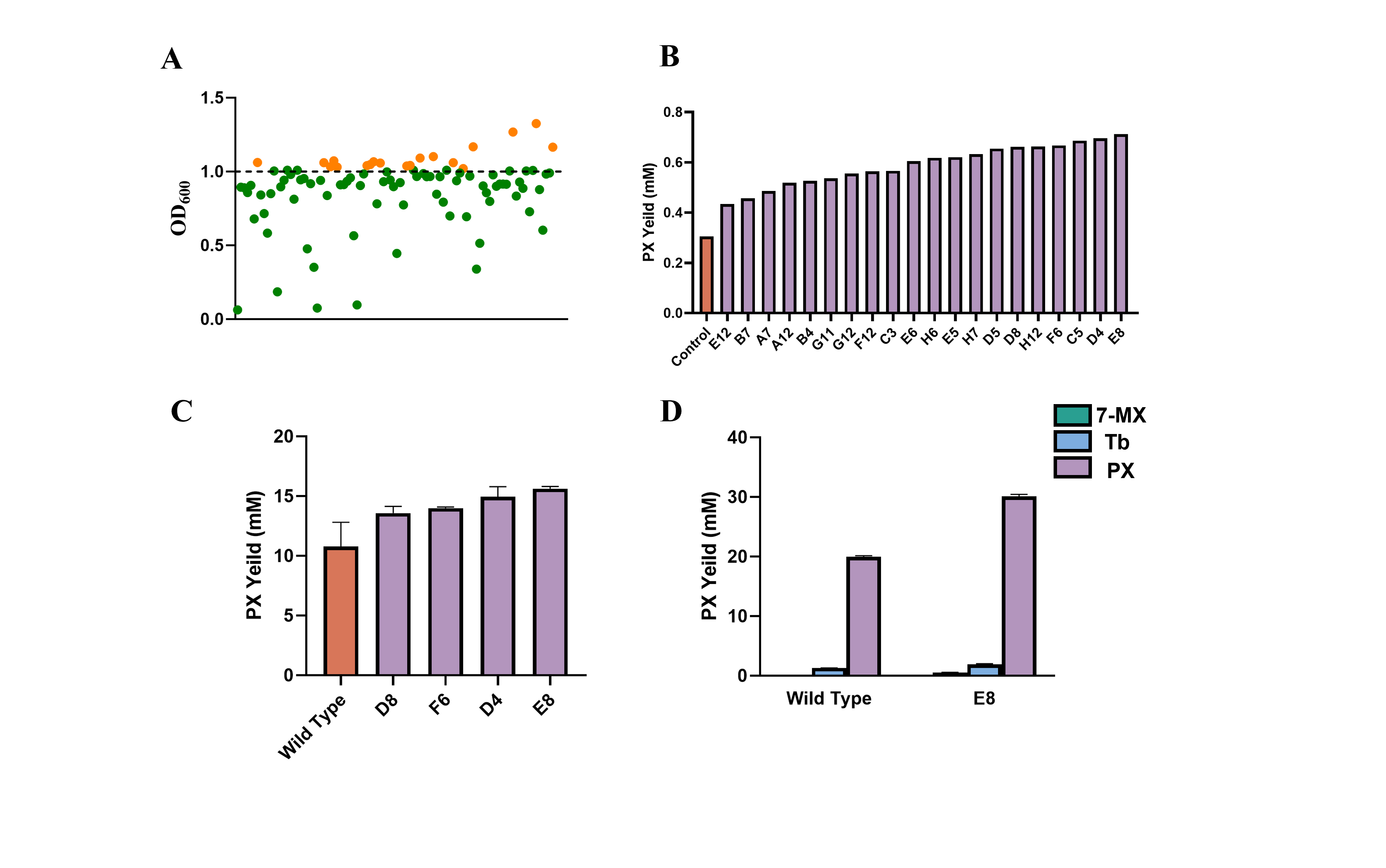
Figure 10. Promoter -10 region mutant screening and PX yield analysis. (A) Screening results of the promoter -10 region mutants. The orange bars indicate strains with OD600 values higher than 1.0. Due to using NdmBQ289T as a template, the overall OD600 values were generally higher during the screening. (B) Twenty strains with OD600 values greater than 1.0 were selected, and their PX yields were measured using HPLC. (C) Further large-scale culture experiments were conducted on four outstanding mutants (D8, F6, D4, E8). Under conditions with OD600 of 100 and a substrate concentration of 20 mM, the PX yield of E8 reached 15.62 ± 0.13 mM, representing an approximately 45% increase compared to the original NdmBQ289T. (D) Under conditions with OD600 of 200 and a substrate concentration of 40 mM, the PX yield of the E8 strain further increased to 30.11 ± 0.23 mM, a 50% improvement over the original NdmBQ289T.
1. Wu, Z., Kan, S. B. J., Lewis, R. D., Wittmann, B. J., & Arnold, F. H, Machine learning-assisted directed protein evolution with combinatorial libraries.Proceedings of the National Academy of Sciences, 116(18), 8852–8858, (2019).
DOI: https://doi.org/10.1073/pnas.1901979116
2. Ishida, T, Effects of Point Mutation on Enzymatic Activity: Correlation between Protein Electronic Structure and Motion in Chorismate Mutase Reaction. Journal of the American Chemical Society, 132(20), 7104–7118, (2010).
DOI: https://doi.org/10.1021/ja100744h
3. Zhou, C., Ye, B., Cheng, S., Zhao, L., Liu, Y., Jiang, J., & Yan, X, Promoter engineering enables overproduction of foreign proteins from a single copy expression cassette in Bacillus subtilis. Microbial Cell Factories, 18(1), 111–121,(2019).
DOI: https://doi.org/10.1186/s12934-019-1159-0