

- De novo rational design: By integrating RFdiffusion and AlphaFold3, we successfully generated IL-2 mimetics that selectively bind IL-2Rβ/γ while sparing IL-2Rα, thereby overcoming the intrinsic limitations of native IL-2 therapy associated with Treg expansion and systemic toxicity.
- Acid-responsive engineering: Incorporation of the pH-sensitive GALA peptide enabled selective IL-2Rβ/γ oligomerization under acidic tumor microenvironments, conferring conditional activation exclusively in low-pH settings.
- Enhanced effector cell potency: The engineered acid-responsive IL-2R agonists markedly promoted NK and CAR-NK cell proliferation, activation, and effector molecule secretion, achieving superior anti-tumor cytotoxicity compared with native IL-2.

1. Developing peptide for selectively binding of IL-2Rβ and IL-2Rγ via de novo design
Based on the natural binding sites between IL-2 and IL-2Rβ and γ (Fig. 1a), we employed RFdiffusion to perform de novo design, generating 100 mimics with high affinity for IL-2Rβ or γ but low affinity for IL-2Rα respectively, each with a length ranging from 46-65 amino acids (Fig. 1b and Extended Data Table 1 and 2). Selection was based on the principle that lower docking scores indicate higher binding affinity. To avoid high affinity for IL-2Rα, sequences exhibiting docking scores below -218 with IL-2Rβ, yet above -215 with IL-2Rα, including B0, B34, and B51 were selected (Extended Data Table 3). Meanwhile, sequences with scores lower than -240 against IL-2Rγ but higher than -245 against IL-2Rα, namely G9, G16, G26, G32, G35, G42, G57, G63, G71, G88, and G90, were included (Extended Data Table 4).
Through visual inspection with PyMOL to obtain the tightly packed helical bundles with a high likelihood of expression as recombinant proteins, we ultimately selected B0, B34, and B51, along with G9, G16, G35, G42, G57, and G71 based on their binding interface to the IL-2R (Fig. 1c). However, the results indicated that most amino acids primarily contributed to structural stability rather than mediating IL-2R binding. Therefore, candidate sequences were truncated to 20 residues directly involved in binding interface contacts, whereas B34 was retained as a 39-amino acid construct to preserve its dispersed interaction sites (Fig. 1d and Extended Data Table 5 and 6).
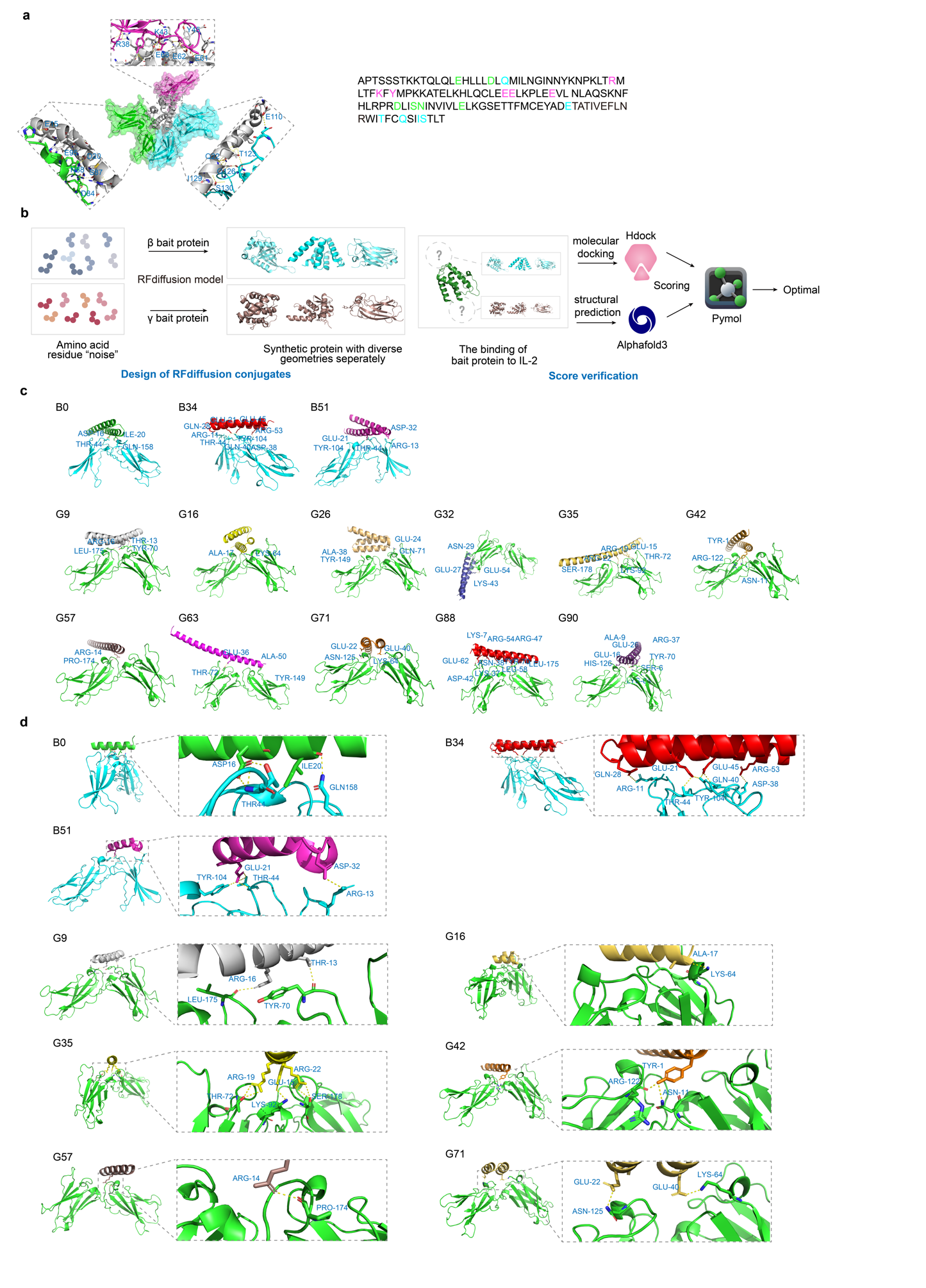
Fig. 1 | De novo design and selection of the IL-2 mimics. a, Structure of IL-2 (cartoon representation) in complex with the IL-2Rαβγ (surface representation) (PDB ID: 2B5I) with their binding interfaces. b, Schematic illustration of the design and score verification process for RFdiffusion conjugates. Amino acid residue “noise” is processed via the RFdiffusion model to generate synthetic proteins with diverse geometries for β and γ bait protein respectively. As for the binding of bait protein to IL-2, molecular docking (via HDOCK) and structural prediction (via AF3) are conducted, followed by scoring to determine the optimal result, which is then visualized through PyMOL. c, The front view of the binding sites between screened mimics with IL-2Rβ and γ separately. d, Binding interfaces between the trimmed sequences and the IL-2Rβ and γ respectively, with the box zooming in to show the amino acids at the binding site (stick representation).
2. Screening and affinity evaluation of IL-2Rβγ agonists
G9, G16, G35, G42, G57, and G71, together with their reverse counterparts, were conjugated via a flexible linker and fused with a glycosylphosphatidylinositol (GPI) anchor to retain IL-2 mimics on the cell surface, while incorporation of a Flag tag enabled subsequent detection and isolation, collectively facilitating NK cell activation (Fig. 2a) Lentiviral transduction of NK-92 cells generated a library of 36 variants, each expressing a distinct IL-2 mimic. To identify the specific sequences that promote NK cell proliferation, the library was seeded into 96-well plates at one cell per well and cultured under cytokine-free conditions. Expanded clones were subsequently harvested and subjected to sequencing (Fig. 2a), which revealed significant enrichment of B34G35R, B51G35R, and B51G9 compared with other constructs (Fig. 2b).
To define the protein-protein interfaces for residue-wise flexibility (B-factor), electrostatic, and van der Waals interaction energy fluctuations, molecular dynamics (MD) simulations were employed to evaluate the affinity of designed IL-2 mimics towards the IL-2Rβγ subunits. Higher RMSD values in the B34G35R complex indicated an unstable receptor binding mode, in contrast to the lower RMSD and enhanced stability observed in the B51G35R complexes (Fig. 2c). The radar plot based on flexibility and conformational entropy features revealed that B51G9 exhibited the lowest conformational diversity and minimal interaction energy fluctuations at the binding interface, suggesting high structural consistency and binding stability, yet a higher B-factor suggested greater atomic uncertainty (Fig. 2d). By contrast, B51G35R showed consistently lower values across all three dimensions, suggesting superior structural stability and identifying it as one of the most promising complexes (Fig. 2d). Bio-layer interferometry (BLI) characterization of binding to IL-2Rα and IL-2Rβγ immobilized on Octet sensor chips revealed KD values separately. In the screening for mimics with low IL-2Rα but high IL-2Rβγ affinity, B51G35R demonstrated the weakest binding to IL-2Rα, substantially below IL-2 (Fig. 2e), while demonstrating the highest affinity toward IL-2Rβ and IL-2Rγ among all tested variants (Fig. 2f). Collectively, these findings establish B51G35R as a selective IL-2Rβγ agonist that minimizes IL-2Rα interaction while preserving high-affinity binding to IL-2Rβγ.
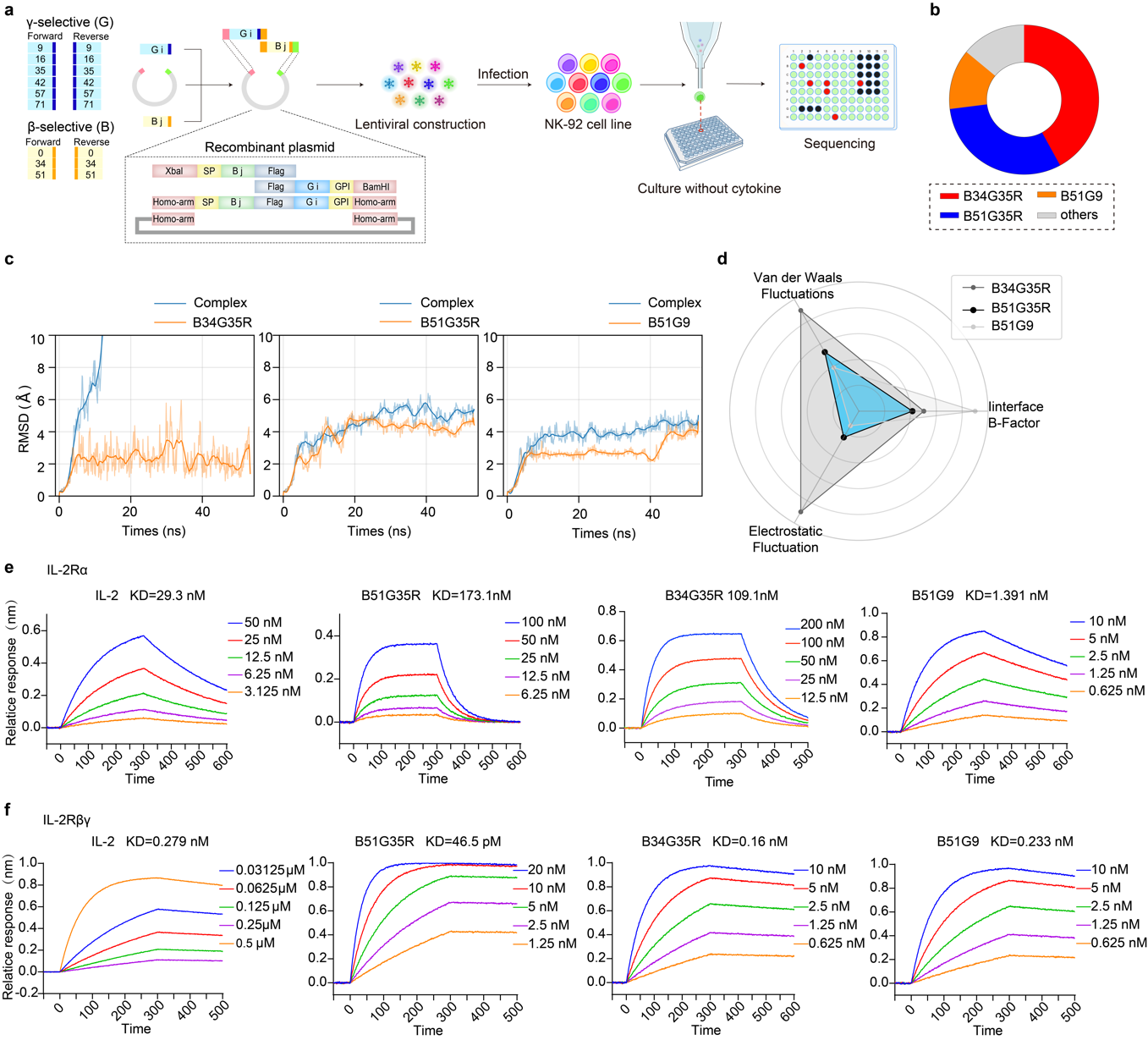
Fig. 2 | Proliferation activity of the eCAR NK-92 cells. a, Construction of four eCARs targeting different antigens (CD19, CD20, GPC3 and MSLN). b-f, Proliferation activity of eCAR NK-92 cells in response to tumor cells stimulation. Tumor cells treated with mitomycin C, including Raji (20 µM), K562(20 µM), HepG2 (30 µM) and AsPC-1 (30 µM). Data are presented as mean ± SD of five independent biological replicates. g, eCAR NK-92 cell activation pathway of JAK/STAT and MAPK indicated by Western Blot.
3. Selective IL-2Rβγ agonists modulate functions of effector cells
After confirming their binding affinity to IL-2R, we systematically evaluated B34G35R, B51G35R, and B51G9 for their capacity to enhance NK cell proliferation, activation, and cytotoxicity, thereby identifying the most effective IL-2 mimics. Among these, B51G35R demonstrated superior potency in driving NK cell expansion compared with native IL-2 (Fig. 3a), and its advantage was further validated in YT cells (Fig. 3b). We further armed αMSLN CAR-NK and αHER2 CAR-T cells with the IL-2 mimics and measured their tumor-killing activity. The heatmap showed that cells expressing B51G35R achieved optimal killing efficacy, outperforming the other two constructs. In contrast, B51G9 only marginally enhanced cytotoxic activity (Fig. 3c).
Effector cell activation and effector molecule secretion were concurrently evaluated. NK cells stimulated with B51G35R displayed markedly enhanced activity, as indicated by increased expression of the degranulation marker CD107a and the activation marker CD69 (Fig. 3d-e). Both B51G9 and B51G35R stimulation significantly increased perforin (PFN) production compared with IL-2. Notably, B51G9 induced substantially higher granzyme B (GrB) secretion than B51G35R, suggesting distinct functional profiles between the mimics (Fig. 3f).

Fig. 3 | Proliferation and in vitro antitumor effect of the screened sequences. a-b, Proliferation activity of NK-92 cells and YT cells in response to B34G51, B34G35, B51G9 and IL-2 stimulation. Data are presented as mean ± SD of three independent biological replicates (*P < 0.05). c, The cytotoxic capabilities of αMSLN CAR-NK and αHER2 CAR-T expressing IL-2 mimics. DMSO was utilized as positive control. d-e, CD107a and CD69 expression were analyzed in effector cells which equipped with IL-2 and its mimics. Data are representative of three independent experiments (**P < 0.01, *P < 0.05). f, The PFN and GrB expression levels were assessed by flow cytometry. Data are representative of at least three independent experiments (**P < 0.01, *P < 0.05).
4. Design and affinity characterization of acid-responsive IL-2 Mimics
To address the limitation that IL-2 fails to promote receptor oligomerization under acidic conditions, the linker in B34G35R, B51G35R, and B51G9 were substituted with GALA4, GALA5, or GALA6 (Fig. 4a). The GALA peptide is a synthetically designed, pH-sensitive motif that transitions from a random coil to an amphipathic α-helix as the pH decreases from 7.0 to 5.0, a property anticipated to drive receptor oligomerization specifically in acidic environments (Fig. 4b). Using AF3, we first assessed the binding interfaces between IL-2Rβγ and the GALA-modified mimics. The results demonstrated that incorporation of the GALA peptide neither compromised structural positioning nor interfered with receptor-binding interactions. Guided by AF3 predicted TM-score (ptm) and inter-residue predicted TM-score (iptm), the GALA5 variant was selected for further experimental validation (Extended Data Table 7). The resulting constructs incorporating GALA5 as a linker are hereafter referred to as B34G35R-G, B51G35R-G, and B51G9-G (Fig. 4c). Subsequent BLI assays under acidic conditions (pH 6.4, acidified with 15 mM lactate) revealed that all three mimics exhibited higher IL-2Rβγ affinity than IL-2, with B34G35R-G (52 pM) displaying substantially stronger binding than the other variants, indicating that GALA-modified IL-2 mimics possess acid-responsive binding capability (Fig. 4d).
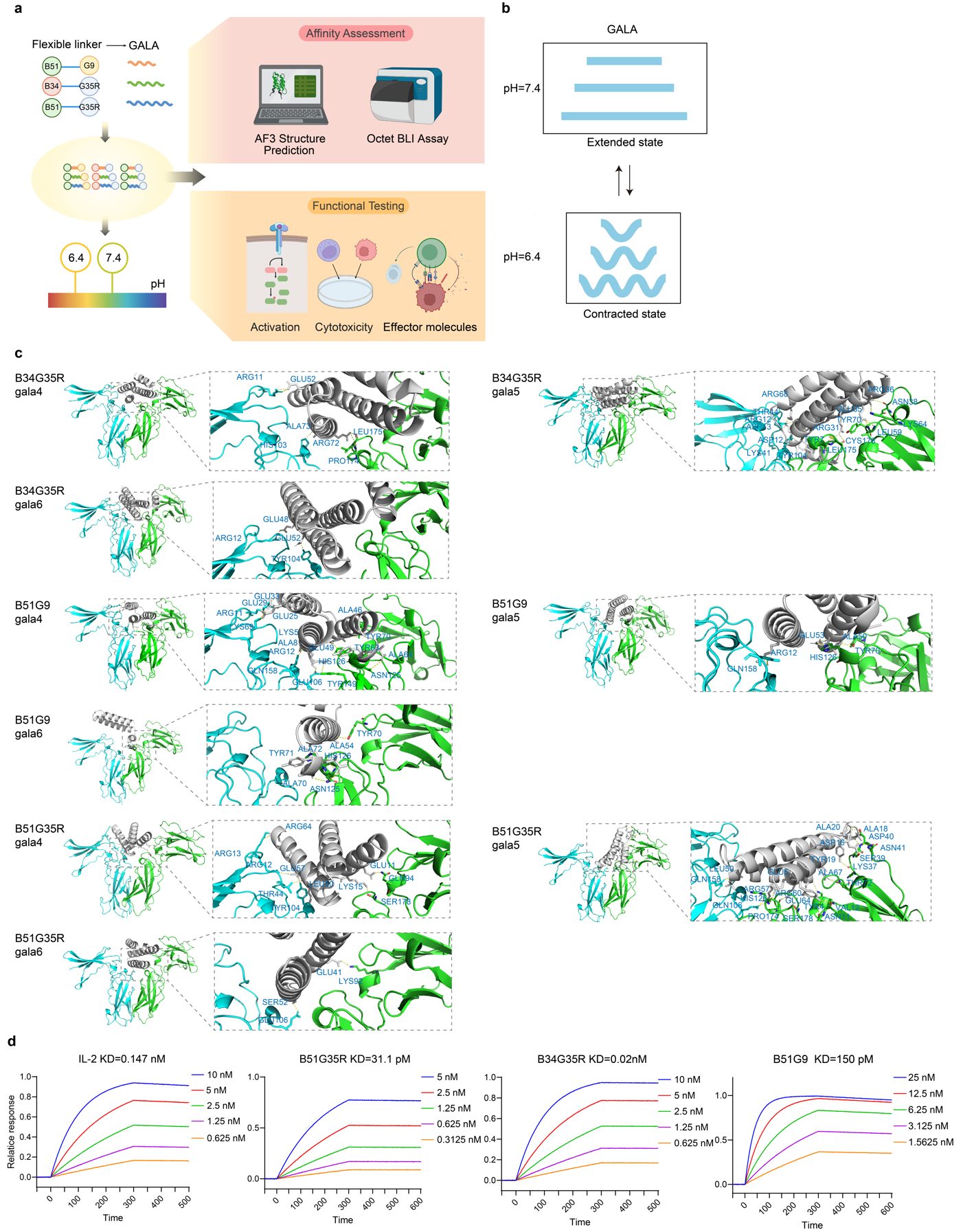
Fig. 4 | GALA replacement and detection of binding affinity under acidic condition. a, Schematic overview of the replacement of GALA, affinity assessment through AF3 structure prediction and BLI assay, and functional testing including activation, cytotoxicity and effector molecules of the designed mimics with where the flexible GGGGS peptide was substituted with GALA4, GALA 5, GALA 6 (4, 5, and 6 represent the number of EALA repeat units in the GALA peptide). b, General depiction of the conformational transition of GALA between extended state at pH=7.4 and contracted state at pH=6.4. c, The front view of the binding sites between B34G35R-G, B51G35R-G, and B51G9-G with IL-2Rβγ, with the box zooming in to show the amino acids at the binding site (stick representation). d, BLI characterization of the binding of B34G35R-G, B51G35R-G, and B51G9-G to IL-2Rβγ.
5. Impact of acid-responsive IL-2R agonists on effector cell receptor oligomerization and functional potency
We evaluated the efficiency of IL-2Rβ/γ oligomerization induced by GALA-linked IL-2 mimics under acidic conditions using the NanoBiT protein complementation assay where the LargeBit and SmallBit fragments were fused to IL-2Rβ and IL-2Rγ, respectively (Fig. 5a). Upon stimulation with the IL-2, B34G35R-G, B51G35R-G and B51G9-G respectively, NanoBiT complementation signals revealed pronounced co-clustering of IL-2Rβ and IL-2Rγ at the plasma membrane compared with IL-2 stimulation alone, visualized as distinct fluorescent puncta. These findings demonstrate that B34G35R-G, B51G35R-G, and B51G9-G, effectively promote IL-2R oligomerization under acidic conditions (pH=6.4) (Fig. 5b).
We evaluated the ability of B34G35R-G, B51G35R-G, and B51G9-G to activate effector cells under different pH conditions. At physiological pH (7.4), B34G35R, B51G35R, and B51G9 strongly induced STAT5 phosphorylation, whereas B51G9-G triggered only mild IL-2R signaling in NK-92 cells, and neither B34G35R-G nor B51G35R-G elevated STAT5 phosphorylation (Fig. 5c, left). In contrast, under acidic conditions (pH 6.4), B34G35R-G and B51G35R-G induced robust STAT5 phosphorylation in NK cells, whereas unmodified constructs completely lost activity (Fig. 5c, right).
Assessment of NK cell expansion revealed that at physiological pH (7.4), the engineered variants B34G35R-G, B51G35R-G, and B51G9-G failed to enhance the tumor-killing efficacy of NK-92 cells. Notably, their activity was exclusively triggered under acidic conditions (pH 6.4). Among them, B51G35R-G exhibited the most potent proliferative capacity, markedly surpassing B34G35R-G and B51G9-G. In sharp contrast, the proliferative activity driven by the non-GALA-modified variants and IL-2 was abolished at pH 6.4 (Fig. 5d). We next evaluated the anti-tumor activity of αMSLN CAR-NK cells expressing these acid-responsive IL-2R agonists. Critically, only under an acidic environment did B34G35R-G and B51G35R-G substantially enhance NK cell cytotoxicity, significantly exceeding their non-GALA-modified counterparts (Fig. 5e). Consistently, changes in CD107a expression as well as PFN and GrB production mirrored these trends (Fig. 5f-h). Collectively, these findings demonstrate that B34G35R-G, B51G35R-G, and B51G9-G are acid-responsive IL-2R agonists capable of significantly enhancing effector cell cytotoxicity under acidic conditions.
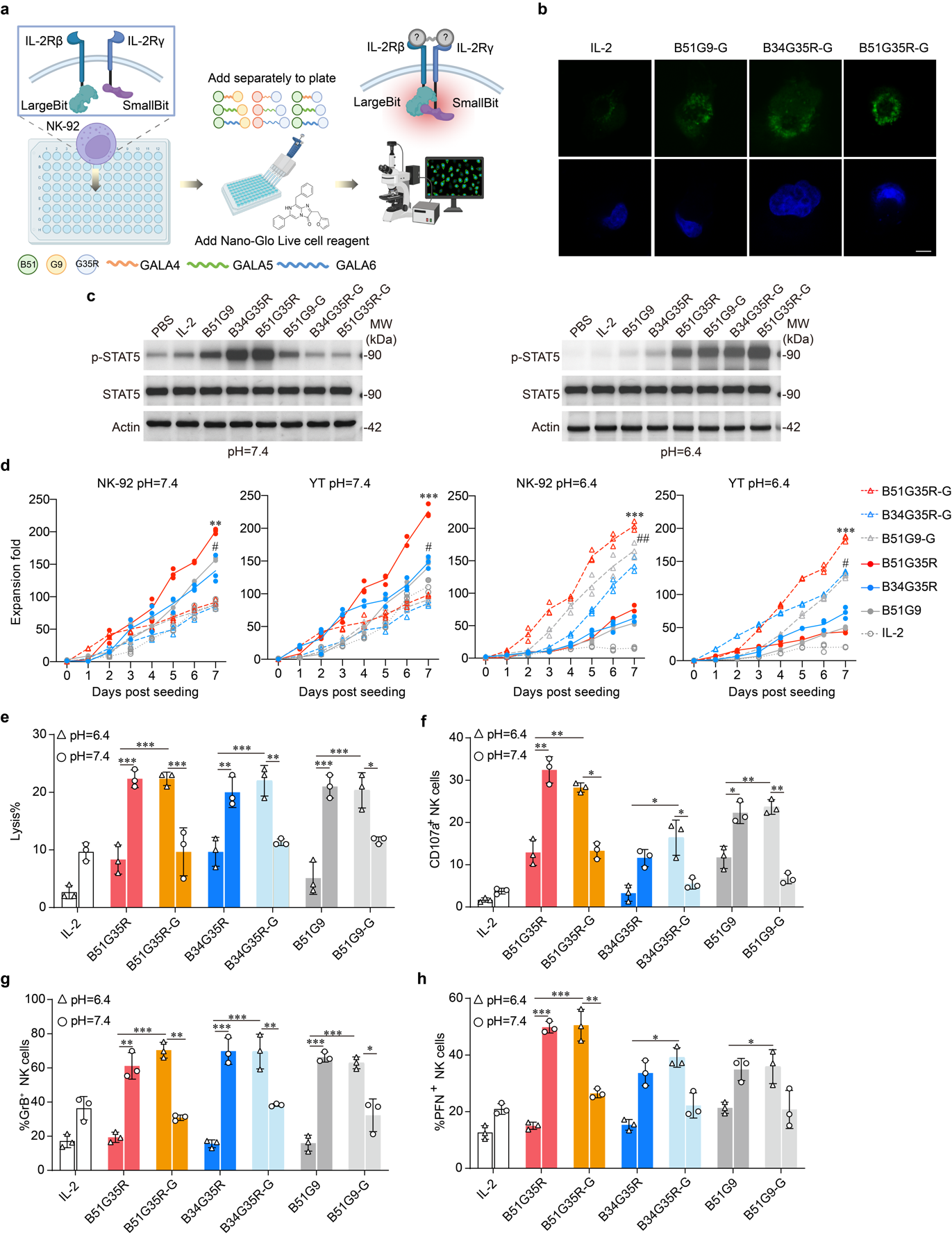
Fig. 5 | Effects of B34G35R-G, B51G35R-G, and B51G9-G on NK cell proliferation and in vitro anti-tumor activity. a, Illustration of the NanoBiT protein complementation assay. The oligomerization induced by fusing LargeBit and SmallBit to IL-2Rβ and IL-2Rγ was verified through immunofluorescence. b, Fluorescence microscopy was utilized to capture images of the NK cells. The green fluorescence indicated the NK-92 cells and the blue fluorescence indicated DAPI, scale bar = 20 μM. c, Expansion fold of NK-92 cells and YT cells at pH=7.4 and pH=6.4 over days post seeding for different constructs (B51G35R-G, B34G35R-G, B51G9-G, B51G35R, B34G35R, B51G9, IL-2). Data are presented as mean ± SD of three independent biological replicates. d, LDH tests for Lysis detection of NK cells expressing IL-2, B51G35R, B51G35R-G, B34G35R, B34G35R-G, B51G9, B51G9-G separately at both pH=6.4 and pH=7.4. Data are representative of at least three independent experiments. e-g, Flow cytometry for CD107a, PFN, GrB detection of NK cells with IL-2 and IL-2 mimics separately at both pH=6.4 and pH=7.4. Data are representative of at least three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001).
