
Shaoteng Lu, Haofeng Yin, Xibo Ye, YuchenGuo, Siqi Yang, Jiachen Zheng, Siyi Wang, Qianshan Hou, Chunzhen Li, Sheng Xu, TianliangLi, Shulei Yin, Yizhi YuNKLII-Evolution-ChinaNational Key Laboratory of Immunity andInflammation, Navy Medical University
The efferocytosis of tumor cells by tumor-associated macrophages (TAMs) leads to an immunosuppressive environment and tumor immune escape. Gas6 is a bridge protein between macrophages and tumor cells. How to modify Gas6 to change the immunosuppressive environment needs to be further explored. In this study, we have developed a novel recombinant fusion protein named Gas6-Fc, which retains the N-terminal domain of Gas6 for binding to apoptotic cells while substituting the C-terminal domain responsible for MerTK receptor binding with an antibody Fc fragment. By engaging with FcR instead of MerTK receptors on macrophages, Gas6-Fc promotes macrophage activation and triggers antibody-dependent cellular phagocytosis (ADCP) towards apoptotic cells. We aim to establish a eukaryotic expression system for the Gas6-Fc fusion protein and enhance its affinity towards tumor cells through directed evolution strategies, enabling it to effectively compete with endogenous Gas6. Subsequently, we will evaluate the impact of Gas6-Fc at both cellular and animal levels. The anticipated outcome is that the Gas6-Fc fusion protein will mitigate the immunosuppressive state within the tumor microenvironment and augment the efficacy of tumor therapies.
Gas6-Fc,Efferocytosis, Macrophage, Cancer, Directed evolution.
Phagocytes, such asmacrophages, play a crucial role in the recognition and engulfment of apoptoticcells, a process known as 'efferocytosis'[1]. Thisprocess is essential for maintaining tissue homeostasis under normalphysiological conditions and facilitating the restoration of homeostasisfollowing disease[2]. Efferocytosis efficiently removes apoptotic cells toprevent their secondary necrosis, which could lead to the release ofpotentially toxic intracellular contents and subsequent inflammation[3,4]. Moreover,phagocytes involved in efferocytosis undergo phenotypic changes towards a morepro-resolution state by increasing the secretion of anti-inflammatory mediatorsand upregulating pro-repair transcriptional programs[4,5]. This remarkablemechanism allows for the removal of billions of dying cells from the bodywithout inducing inflammation or immune responses. In contrast toefferocytosis, other forms of phagocytosis such as Fc receptor-mediatedphagocytosis are primarily aimed at initiating an immune response.
Theimmunologically silent process of efferocytosis, however, creates anenvironment for cancer cells to evade immune surveillance and thus promotestumor progression[6,7]. Harsh conditions within the tumor microenvironment,such as starvation, hypoxia, and chemotherapy drugs, induce apoptosis in tumorcells and trigger robust efferocytosis by tumor-associated macrophages.Following efferocytosis, tumor-associated macrophages upregulate the productionof anti-inflammatory cytokines, such as Transforming growth factor (TGF)β andinterleukin (IL)-10[7,8]. Additionally, tumor-associated macrophages expressvarious immune checkpoint ligands including programmed death-ligand 1 (PD-L1),leading to cytotoxic CD8 T cell starvation through arginase expression anddepletion of essential amino acids[9,10]. Furthermore, tumor-associatedmacrophages recruit regulatory T cells (Tregs), which contribute to theinhibition of antitumor immune responses by suppressing pro-inflammatorycytokine production and effector T cell function[11,12]. In this context, theaccumulation of tumor-associated macrophages is associated with an unfavorableprognosis in several cancer types including melanoma, breast, pancreatic, liver,ovarian, head and neck, bladder and renal cell cancer[10,13,14].
Growtharrest-specific 6 (Gas6), a 75kDa vitamin K-dependent secreted protein, servesas a critical regulator of efferocytosis by acting as bridging proteins[15].The N-terminal GLA domain (γ-carboxyglutamate domain) of Gas6 specificallybinds to phosphatidylserine (PS) located on the surface of apoptotic cells,while the C-terminal region interacts with the MerTK receptor present onmacrophage surfaces[16]. This bridging effect facilitates the recognition,phagocytosis, and clearance of apoptotic cells by macrophages and inducesimmunosuppression in macrophages[15,17]. In the tumor microenvironment, thesustained activation of GAS6/MerTK signaling pathway leads to the generation ofa series of immunosuppressive signals that inhibit the host's anti-tumor immuneresponse[18]. Consequently, this pathway mediates tumor drug resistance andimmune evasion, making it one of the most promising next-generation therapeutictargets for tumors[19–21].
TheFc fusion protein refers to a genetically engineered protein that combines abiologically active protein segment with the Fc fragment in immunoglobulin[22].This fusion not only retains the specific function of the original protein butalso incorporates the functional characteristics of the Fc domain, making it avaluable therapeutic tool for various diseases, including cancer[23]. Comparedto the original protein, the Fc fusion protein exhibits an extended half-lifeand enhanced stability due to disulfide bond formation within its dimericstructure facilitated by the Fc domain[24,25]. Additionally, the Fc domain canmediate tumor-killing effects such as inflammation, antibody-dependent cellcytotoxicity (ADCC), and antibody-dependent cell phagocytosis (ADCP)[26,27]. Inthis study, we have successfully engineered, expressed, and purified theGas6-Fc fusion protein with the aim of enhancing the immunosuppressive tumormicroenvironment. In vitro experiments were conducted to validate thefunctionality of the fusion protein in specifically targeting apoptotic cellsand improving the immunosuppressive tumor microenvironment. Directed evolutionhas been employed to enhance the affinity of the fusion protein towardsapoptotic cell targets.
Gas6 comprises the γ-carboxyglutamicacid (Gla) domain, four epidermal growth factor (EGF)-like domains, and twolaminin G-like (LG) domains (Fig. 1A). Considering the immunosuppressivefunction of Gas6 and the potent immune activation function of Fc, we retainedthe Gla domain and the EGF-like domain, which bind Ca2+ to fulfillthe Ca2+ dependency of Gla functioning, and fused them with the Fcfragment using a flexible linker (Fig. 1B). Utilizing this protein design approach, wegenerated a eukaryotic expression plasmid for Gas6-Fc by optimizing pcDNA3.1vector with a flag tag integration (Fig. 1C). The gene sequence encodinghGas6 (GenBank accession no. NM_008202.4) was modified to retain its signalpeptide as well as Gla and EGF regions while also being tailored to enhancehost preference based on sequence alterations illustrated in Fig. 1D. Wepropose that linking Gas6-Fc between macrophages and tumor cells could triggeractivation signals while alleviating immunosuppression (Fig. 1E). Thewild-type Gas6 binds to apoptotic cells and initiates immunosuppressivesignals, leading to the polarization of macrophages towards the M2 phenotype,secretion of anti-inflammatory factors such as IL-10, and induction of immunetolerance. The N-terminus of Gas6-Fc competes with the wild-type Gas6 forbinding to PS on the surface of apoptotic cells. On the other hand, theC-terminus of Gas6-Fc triggers immune activation signals, polarizes macrophagestowards the M1 phenotype, and stimulates production of pro-inflammatory factorsincluding IL-1β, IL-18, and IFNβ.
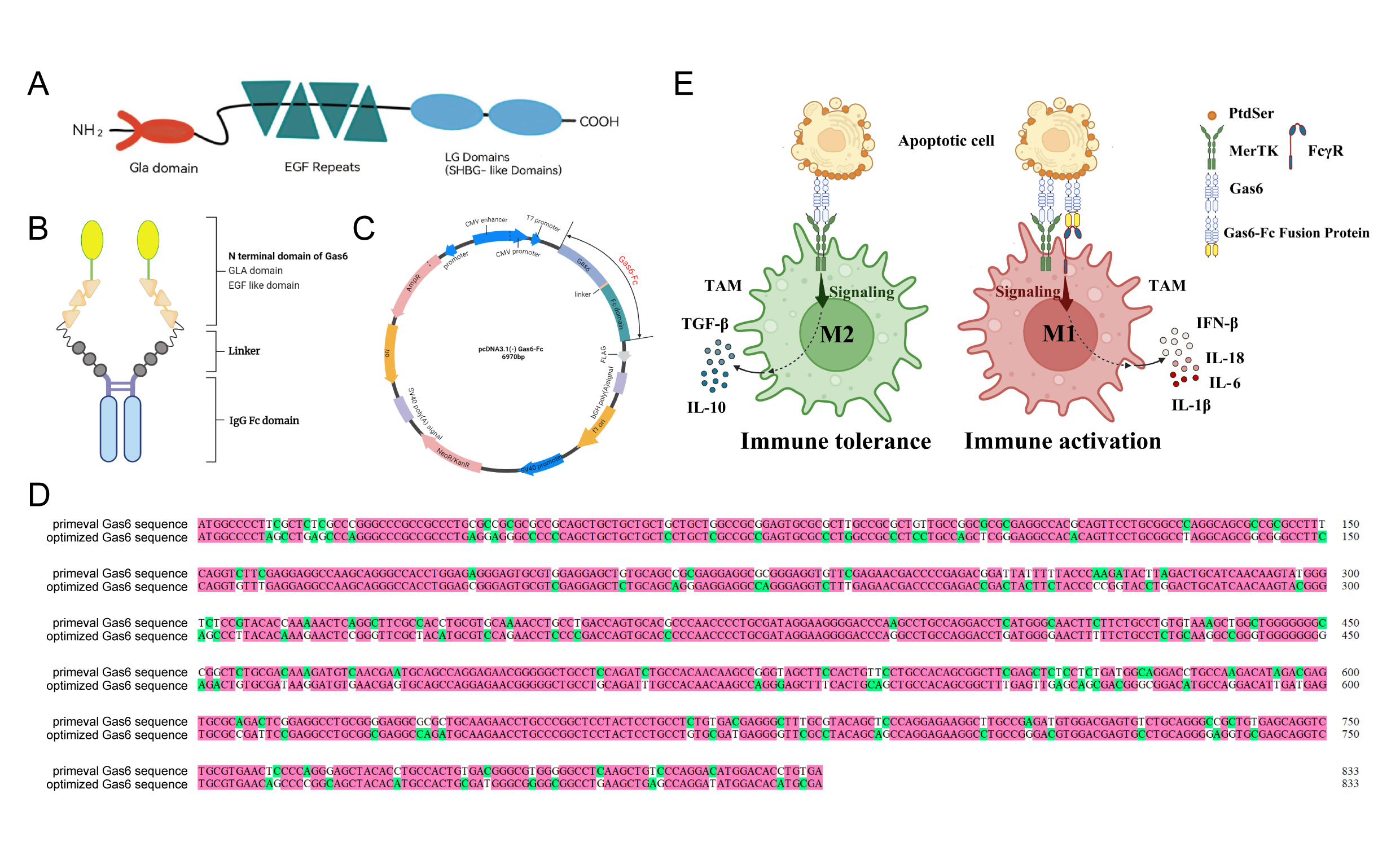
Figure 1. Thedesign strategy of the Gas6-Fc fusion protein. AThe structures of Gas6. B The structures of Gas6-Fc. C Theplasmid map of Gas6-Fc. D Comparative display of sequence changes. ESchematic diagrams of the interaction between apoptoticcell/Gas6-Fc/macrophage.
The pcDNA3.1-Gas6-Fc-Flagplasmid was transfected into HEK-293T cells for 24 hours, and the supernatantand cell lysate from the transfected cells were collected. The proteins in thesupernatant were enriched through anti-Flag immunoprecipitation. In comparisonto the untransfected sample, both the enriched supernatant and cell lysatesamples from HEK-293T cells transfected with Gas6-Fc plasmid exhibited adistinct band at a molecular weight of 63 KD, which corresponded to thepredicted size of Gas6-Fc (Fig. 2A). These results demonstratesuccessful expression of the Gas6-Fc plasmid.
In order to minimize theimpact of serum on isolation and purification (and to reduce costs), weutilized Expi293F cells, which can be cultured in a serum-free medium forlarge-scale protein production, to achieve high-level expression of the Gas6-Fcfusion protein. The Gas6-Fc plasmid was transiently transfected into Expi239Fcells. Subsequently, Gas6-Fc in the cell culture media was purified usingProtein G affinity chromatography (Fig. 2B). Protein samples at various purification stageswere analyzed by 12% SDS-PAGE. Coomassie brilliant blue staining resultsindicated successful protein expression and purification in Expi239F cells (Fig.2C). The results demonstrated successful expression of Gas6-Fc fusionprotein in the culture media of Expi293F cells, followed by its effectiveisolation through protein G affinity chromatography. Furthermore, we performedan additional column repetition to enhance the efficiency of fusion proteinenrichment, resulting in a final yield of 5ml fusion protein at a concentrationof 128ug/ml.
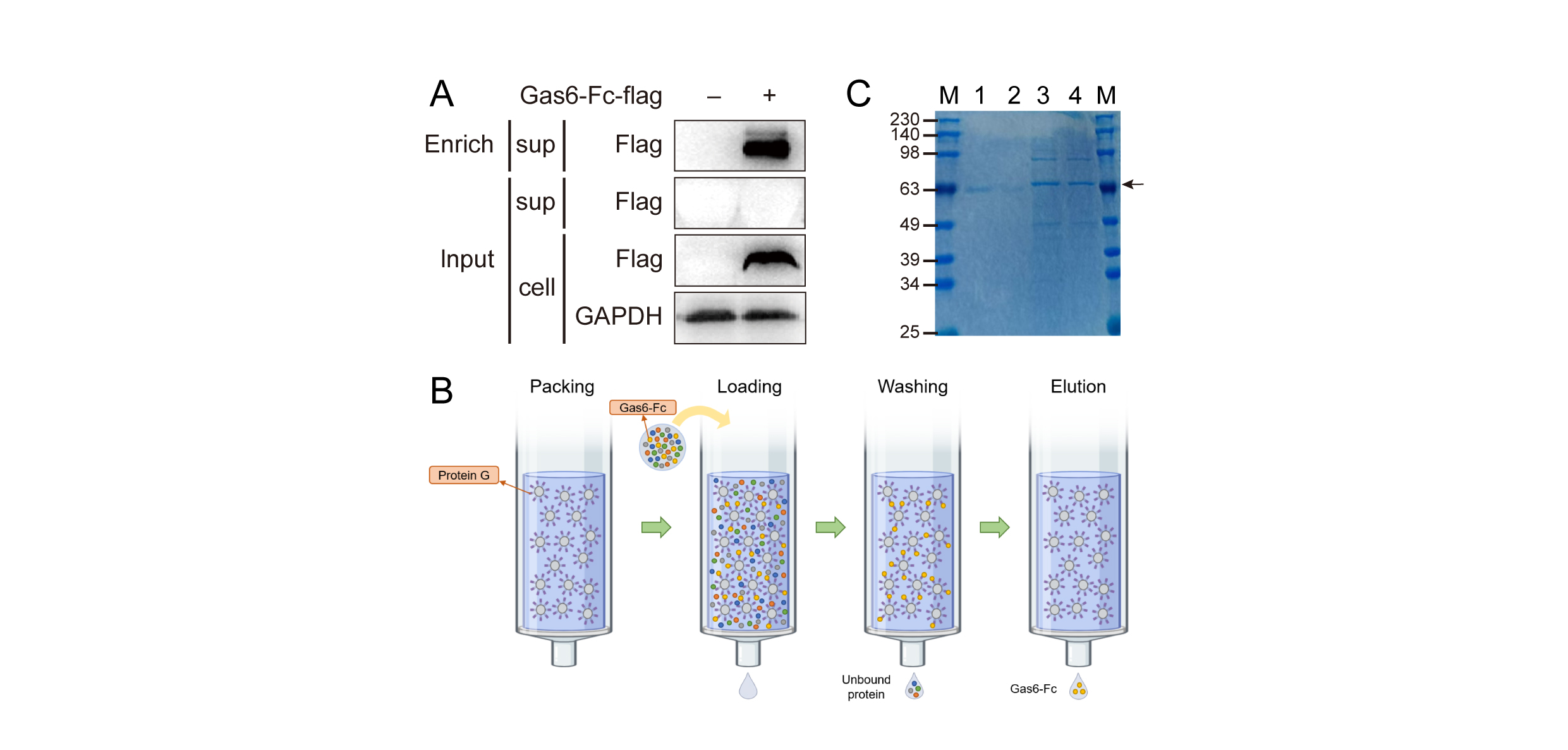
Figure 2. Preparationprocedure for the fusion protein Gas6-Fc. A Protein expression insupernatant and cell lysate of HEK-293T after transfection with Gas6-Fc plasmidfor 24 h. B Flow chart of Gas6-Fc protein purified by protein G affinitychromatography. C SDS-PAGE identification of purified Gas6-Fc protein.M: marker; 1: purified Gas6-Fc; 2: eluted effluent; 3: original solution; 4:after a purification of the original solution.
The ability of theengineered Gas6-Fc fusion protein to specifically target phosphatidylserine(PS) on the surfaces of apoptotic cells was confirmed through flow cytometryanalysis. Human T-lymphocytic leukemia cells (Jurkat) were treated with staurosporine(STS) at a ratio of 250:1 for a duration of 3 hours to induce apoptosis.AnnexinV-FITC, a PS binding protein, was used to detect the percent ofapoptosis cells. After treatment with STS, the percentage of apoptotic Jurkatcells significantly increased from 3.5% to 98.1% (Fig. 3A, B).Subsequently, apoptotic Jurkat cells were incubated with Gas6-Fc or isotypeIgG. The binding of Gas6-Fc to apoptotic cell surfaces was detected using ananti-Fc (PE) secondary antibody. As expected, it was found that IgG did notbind to Jurkat cells either with or without apoptotic treatment (Fig. 3C, D).In contrast, The binding ratio of Ga6 to Jurkat cells exhibited a significantincrease following apoptosis induction, rising from 13.3% to 58.6% (Fig. 3C,D). These findings strongly suggest that the Gas6-Fc fusion proteinexhibits an affinity for PS on the surface of apoptotic cells and caneffectively target them.
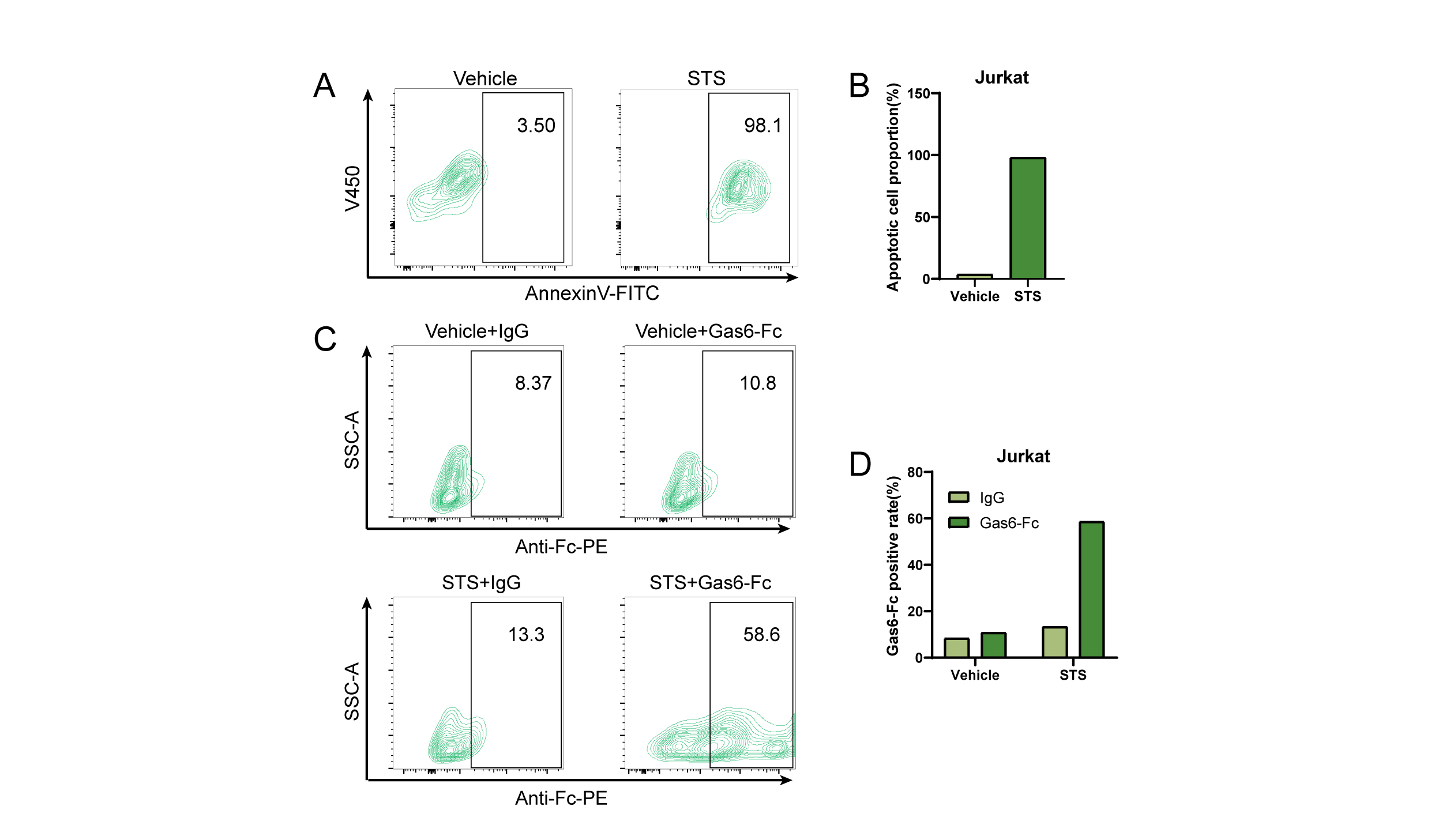
Figure 3. Thetargeting capability of GAS6-Fc towards apoptotic cells. A Flow cytometryquantification of apoptotic Jurkat cells with or withoutSTS treatment. B Rate of apoptotic Jurkat cells with or withoutSTS treatment. C The binding rate of Gas6-Fc to Jurkat cells wasquantified using flow cytometry, with detection by an anti-Fc (PE) antibody. DPositive rate of Jurkat cells binding to Gas6-Fc.
In order to validate the functionalityof Gas6-Fc fusion protein in inducing immune activation of macrophages, wecultured macrophages with Gas6-Fc fusion protein in a culture medium containingapoptotic Jurkat cells. The presence of apoptotic cells would induce theefferocytosis of macrophages. After a 7-hour incubation period, the RNA ofmacrophages was extracted and cytokine expression was assessed using qPCR. Thelevels of immune-activating cytokines, including IL6 and IFN-β,increased significantly with 100ug/ml Gas6-Fc administration duringefferocytosis compared to the control group without Gas6-Fc treatment (Fig.4A), indicating Gas6-Fc can enhance the secretion of pro-inflammatorycytokines. In contrast, administration of Gas6-Fc resulted in a decrease in theexpression level of IL10, an immunosuppressive cytokine, in macrophagescompared to the control group (Fig. 4B). 1. Overall, these data suggestthat Gas6-Fc can shift macrophages from an immunosuppressive state to an immune-activatingstate during efferocytosis.
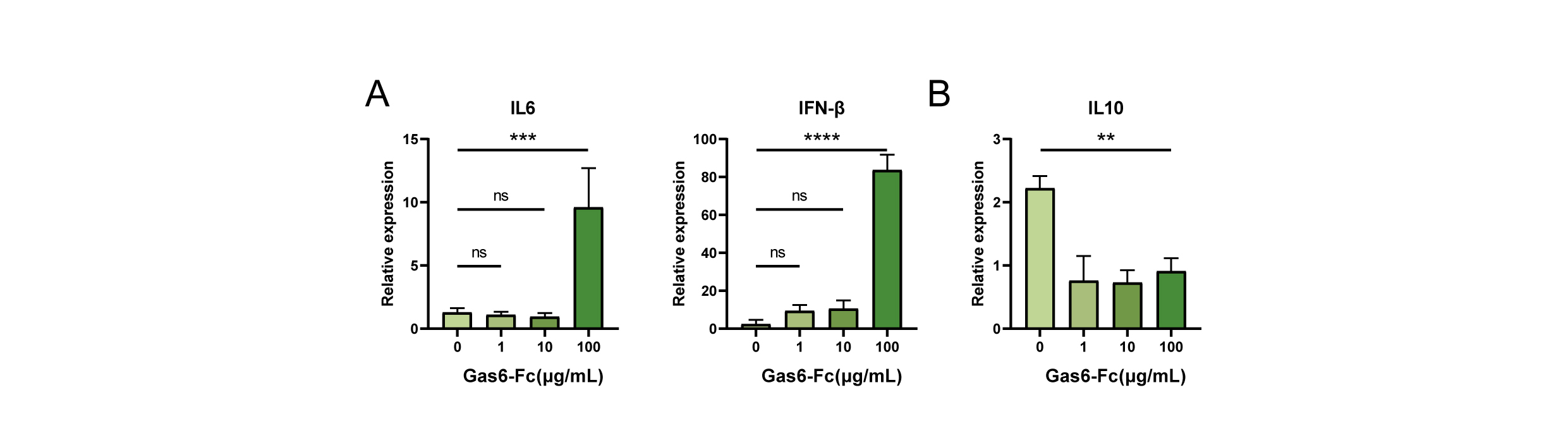
Figure 4. The Gas6-Fc fusion proteinalleviates immune suppression of macrophages.A The RNA levels of IL6 and IFN-β in macrophages treated with Gas6-Fcfusion protein. B The RNA level of IL10 in macrophages treated withGas6-Fc fusion protein. 1 μg/ml, 10 μg/ml, and 100 μg/ml are the threeconcentration gradients of Gas6-Fc fusion protein in the system. The datarepresent three independent replicate experiments.
Considering that only ahigh concentration (100ug/ml) of Gas6-Fc exhibited an ameliorated immunesuppression effect, and taking into account the in vivo experimentaldosage as well as clinical drug requirements, our objective is to furtherinvestigate methods for optimizing the fusion protein's affinity to PS on apoptoticcells. As, the Gla and EGF domains of Gas6 are crucial regions for binding toPS, we introduced random mutations into these two regions by error-prone PCR. Inspiredby the epitope localization function of phage display technology, weestablished a phage peptide display library targeting the mutated Gla and EGFfragments (Fig. 5A). The phage display procedure was as follows: Phagedisplay library generation: The gene fragments with random mutations wereobtained using error-prone PCR, followed by enzymatic ligation for theirinsertion into phasmid vectors. The library phasmids were then transferred intoE. coli via electroshocking to establish a bacterial library. Helper phage wasintroduced to invade the E. coli cells, facilitating the amplification of phagewithin the bacterium and ultimately constructing a recombinant phage librarycapable of displaying mutated Gla-EGF fragment protein on its surface. (2)Targeted binding: Gla-EGF fragment protein-display phages were incubated with magneticnanoparticles loaded with PS. (3) Washing: We eliminated the phages thatexhibit non-binding or weak binding interactions with PS-loaded nanoparticles,and specifically isolate the phages with high affinity. (4) Phage Elution: Thehigh affinity phage was eluted for subsequent sequence analysis. (5) Phagere-amplification: Take a small amount of phage to reinfect E.coli and introducehelper phage for proliferation, in preparation for another enrichment. Afterseveral cycles, collect purified phagemids for analysis. Randomly mutated Gla-EGFfragments were generated using the GeneMorphII random mutagenesis kit. The fragmentswere inserted between the SacI and SpeI enzyme cleavage sites adjacent to the5' end of M13 gene PIII, resulting in a genetically engineered constructsuitable for displaying the corresponding protein fragments (Fig. 5B). Thedisplayed protein in our system is tagged with an HA tag, and its recognitionserves as confirmation for the presence of each individual Gla-EGF fragment onthe phage surface (Fig. 5B). The analysis of the double digestionproduct of the phagemid vector pcomb3xss using 1% agarose gel electrophoresisrevealed two distinct bands that corresponded to the expected size (3300 bp +1600 bp) (Fig. 5C). The gel electrophoresis analysis revealed thepresence of mutated Gla-EGF fragments generated through error-prone PCR, withan expected band size ranging between 500 and 750bp (Fig. 5D). Thephages enriched with a high affinity for PS have been submitted for sequencing,pending subsequent functional validation of the high-affinity mutant. Moreover,the antitumor function of the evolved Gas6-Fc would be evaluated through an invivo tumor model,
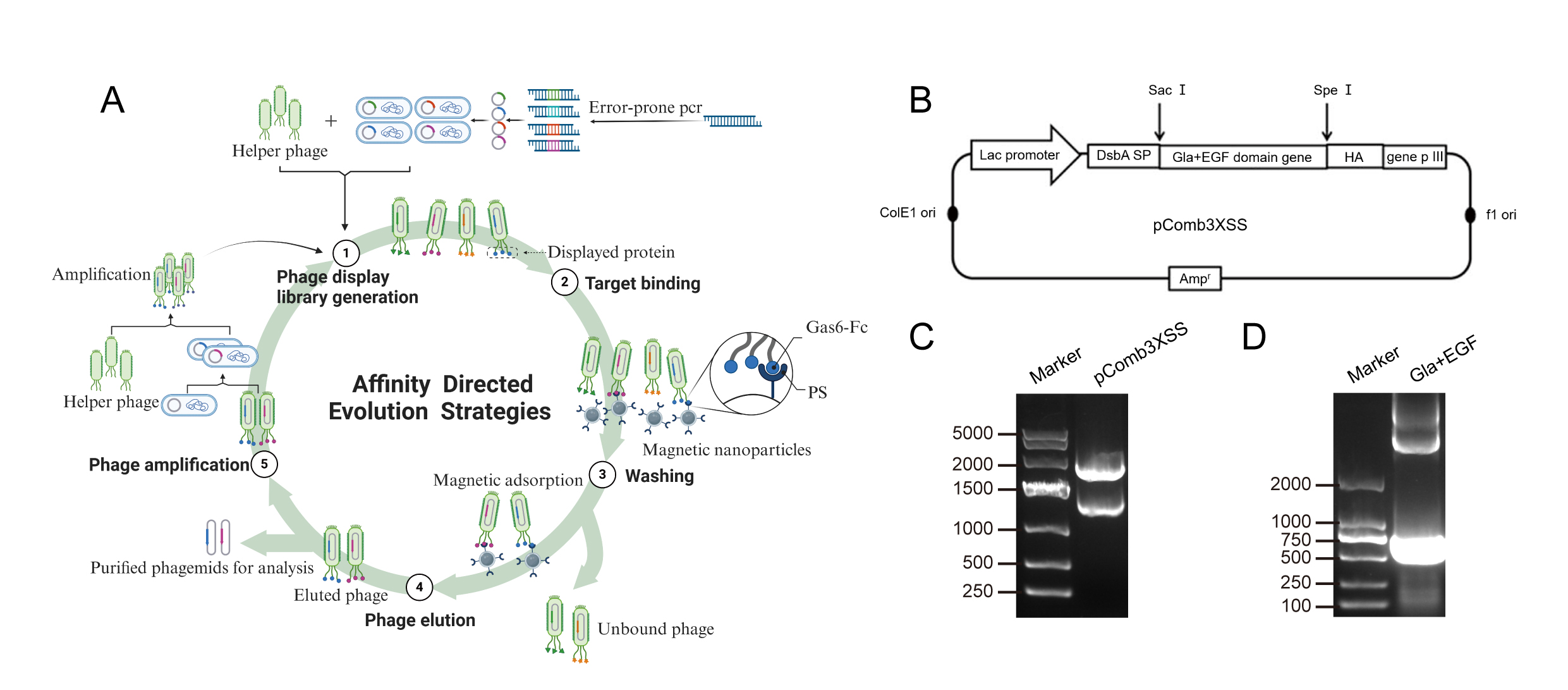
Figure 5. Strategy of affinity directed evolution. A Schematic diagram of phagedisplay technology B Schematic representation of phagemid vector. C Phagemidvector pComb3xss was digested by double enzyme (spe1/sac1) and detected usinggel electrophoresis D The generated Gla-EGF fragments by error-prone PCRwere detected using gel electrophoresis.
Incontrast to wild-type Gas6, the absence of the MerTK binding domain in Gas6-Fcprevents M2 polarization of macrophages. Instead, through its Fc domain bindingto FcγRs, Gas6-Fc promotes M1 polarization by activating the NF-κB signalingpathway and enhancing secretion of IFN-β, IL-18, and IL-1β by macrophages. Consequently,this activation leads to enhanced anti-tumor immune responses. Moreover,Gas6-Fc also activates diverse immune cells involved in antibody-dependentcell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis(ADCP), and complement-dependent cytotoxicity (CDC) effects, thereby furtheraugmenting tumor cell clearance. Engineered through directed evolution, Gas6-Fcexhibits significantly higher affinity for PS than wild-type Gas6 and isexpected to competitively inhibit the binding between wild-type Gas6 andapoptotic cells at low concentrations. While wild-type Gas6 has a smallermolecular weight and is susceptible to hydrolysis by endogenous proteases andpeptidases, Gas6-Fc possesses greater protein stability.
Inorder to enhance the affinity of Gas6-Fc for PS, we introduced random mutationsin a specific region of Gas6 using error-prone PCR to establish a diverselibrary of Gas6 mutants. To enrich for mutants with the highest affinity forPS, we employed phage display technology, known for its cost-effectiveness,high efficiency, and short cycles. The peptide library was displayed on thesurface of M13 phage and incubated with PS-coated magnetic beads duringselection. Subsequently, we enriched for Gas6 mutants exhibiting the strongestbinding affinity towards PS. Furthermore, our future studies will investigatethe potential influence of the Fc segment on the interaction between the Gas6segment and PS.
Inaddition to Gas 6, there exist other PS-binding proteins that can be engineeredinto Fc-fusion proteins to exert therapeutic effects. For instance, Annexin A5is a calcium-dependent phospholipid-binding protein exhibiting high affinitytowards PS... It has been demonstrated that the interaction between Annexin A5and PS on apoptotic cell surfaces can modulate PS-mediated immune suppression,augment the immunogenicity of apoptotic cells, and alleviate immune suppressiveconditions within the tumor microenvironment. Due to its smaller size andnon-secretory nature, Annexin A5's relatively shorter half-life may impacttreatment efficacy or necessitate higher dosages. Consequently, fusion ofAnnexin A5 with the Fc region could potentially prolong its half-life andenhance therapeutic effectiveness.
1 Boada-RomeroE, Martinez J, Heckmann BL, et al. The clearance of dead cells byefferocytosis. Nat Rev Mol Cell Biol. 2020;21:398–414. doi:10.1038/s41580-020-0232-12
2、GerlachBD, Ampomah PB, Yurdagul A, et al. Efferocytosis induces macrophageproliferation to help resolve tissue injury. Cell Metab.2021;33:2445-2463.e8. doi: 10.1016/j.cmet.2021.10.0153
3、DoranAC, Yurdagul A, Tabas I. Efferocytosis in health and disease. Nat RevImmunol. 2020;20:254–67. doi: 10.1038/s41577-019-0240-64
4、PoonIKH, Ravichandran KS. Targeting Efferocytosis in Inflammaging. Annu RevPharmacol Toxicol. 2024;64:339–57. doi:10.1146/annurev-pharmtox-032723-1105075
5、MehrotraP, Ravichandran KS. Drugging the efferocytosis process: concepts andopportunities. Nat Rev Drug Discov. 2022;21:601–20. doi:10.1038/s41573-022-00470-y6
6、LecoultreM, Dutoit V, Walker PR. Phagocytic function of tumor-associated macrophages asa key determinant of tumor progression control: a review. Journal forImmunotherapy of Cancer. 2020;8:e001408. doi: 10.1136/jitc-2020-0014087
7、ZhouY, Yao Y, Deng Y, et al. Regulation of efferocytosis as a novel cancertherapy. Cell communication and signaling: CCS. 2020;18:71. doi:10.1186/s12964-020-00542-98
8、QiuH, Shao Z, Wen X, et al. Efferocytosis: An accomplice of cancer immuneescape. Biomedicine & Pharmacotherapy. 2023;167:115540. doi:10.1016/j.biopha.2023.1155409
9、ZhangH, Liu L, Liu J, et al. Roles of tumor-associated macrophages inanti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. 2023;22:58.doi: 10.1186/s12943-023-01725-x10
10、XiangX, Wang J, Lu D, et al. Targeting tumor-associated macrophages tosynergize tumor immunotherapy. Signal Transduction and Targeted Therapy.2021;6:75. doi: 10.1038/s41392-021-00484-911
11、LiC, Xu X, Wei S, et al. Tumor-associated macrophages: potentialtherapeutic strategies and future prospects in cancer. Journal forImmunotherapy of Cancer. 2021;9:e001341. doi: 10.1136/jitc-2020-00134112
12、PathriaP, Louis TL, Varner JA. Targeting Tumor-Associated Macrophages in Cancer. TrendsImmunol. 2019;40:310–27. doi: 10.1016/j.it.2019.02.00313
13、KumariN, Choi SH. Tumor-associated macrophages in cancer: recent advancements incancer nanoimmunotherapies. Journal of experimental & clinical cancerresearch: CR. 2022;41:68. doi: 10.1186/s13046-022-02272-x14
14、Cheng K, Cai N, Zhu J, et al. Tumor-associatedmacrophages in liver cancer: From mechanisms to therapy. CancerCommunications. 2022;42:1112–40. doi: 10.1002/cac2.1234515
15、WuG, Ma Z, Cheng Y, et al. Targeting Gas6/TAM in cancer cells and tumormicroenvironment. Mol Cancer. 2018;17:20. doi: 10.1186/s12943-018-0769-116
16、WuG, Ma Z, Hu W, et al. Molecular insights of Gas6/TAM in cancerdevelopment and therapy. Cell Death Dis. 2017;8:e2700. doi:10.1038/cddis.2017.11317
17、VagoJP, Amaral FA, van de Loo FAJ. Resolving inflammation by TAM receptoractivation. Pharmacol Ther. 2021;227:107893. doi:10.1016/j.pharmthera.2021.10789318
18、MyersKV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors):implications for macrophages in the tumor microenvironment. Mol Cancer.2019;18:94. doi: 10.1186/s12943-019-1022-219
19、Adam-ArtiguesA, Arenas EJ, Martínez-Sabadell A, et al. Targeting HER2-AXLheterodimerization to overcome resistance to HER2 blockade in breast cancer. SciAdv. 2022;8:eabk2746. doi: 10.1126/sciadv.abk274620
20、Apostolo D, Ferreira LL, Vincenzi F, et al.From MASH to HCC: the role of Gas6/TAMreceptors. Front Immunol. 2024;15:1332818. doi:10.3389/fimmu.2024.133281821
21、ZhuC, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancerapproach: functions, molecular mechanisms and clinical applications. MolCancer. 2019;18:153. doi: 10.1186/s12943-019-1090-322
22、RashidMH. Full-length recombinant antibodies from Escherichia coli: production,characterization, effector function (Fc) engineering, and clinical evaluation. MAbs. 2022;14:2111748. doi: 10.1080/19420862.2022.211174823
23、Niu Y-X, Xu Z-X, Yu L-F, et al. Advancesof research of Fc-fusion protein that activate NK cells for tumorimmunotherapy. Int Immunopharmacol. 2022;109:108783. doi:10.1016/j.intimp.2022.10878324
24、RamdaniY, Lamamy J, Watier H, et al. Monoclonal Antibody Engineering and Designto Modulate FcRn Activities: A Comprehensive Review. Int J Mol Sci.2022;23:9604. doi: 10.3390/ijms2317960425
25、FrommG, de Silva S, Schreiber TH. Reconciling intrinsic properties of activating TNFreceptors by native ligands versus synthetic agonists. Front Immunol.2023;14:1236332. doi: 10.3389/fimmu.2023.123633226
26、YuJ, Song Y, Tian W. How to select IgG subclasses in developing anti-tumortherapeutic antibodies. J Hematol Oncol. 2020;13:45. doi:10.1186/s13045-020-00876-427
27、Musolino A, Gradishar WJ, Rugo HS, et al.Role of Fcγ receptors in HER2-targeted breast cancer therapy. J ImmunotherCancer. 2022;10:e003171. doi: 10.1136/jitc-2021-003171